转座子(TEs)是一种内源移动元件,可以在其宿主基因组内移动并自主或非自主地增加其拷贝数。它们几乎存在于所有已经测序的基因组中,通过不同的结构和移动机制进行表达。TEs的大小范围从小于100 bp(例如MITEs [2])到大于20 kb不等。在哺乳动物基因组中,很大一部分的调控元件结合位点、人类基因组超过一半的开放染色质的区域都被证实是由转座子衍生而来。然而,由于其序列本身的高度的重复性,它们的识别、注释和动态跟踪较为复杂。虽然现在已经开发了许多工具来进行TEs的注释,如REPET,RepeatMasker和EDTA,但均需要大量的计算资源且准确率有待提高。此外,追踪个体间或世代间特定TE插入的有无仍然局限于繁重的湿实验方法。
在过去的15年中,特别是Illumina短读测序技术的进步,使得不仅可以对一个物种或亚种内的个体进行测序,还可以对整个群体进行测序。大规模测序数据的主要发现之一是TEs在基因组中的变化最为明显。因此,已经开发了许多工具使用短的reads数据在样本间(例如T-lex;McClintock)或群体内(例如DNApipeTE;PoPoolationTE2)识别潜在的TE多态性。然而,短的reads数据并不适合结构变异(SV)的鉴定,尤其是由TE插入/删除引起的SV。相反,长读测序技术例如Pacific Biosciences和Oxford Nanopore Technologies(ONT)则可以获得长度大于20kb的reads(ONT甚至可以获得长度大于100kb的reads)。因此,一个单独的reads可能会包含完整的结构变异或TE插入。然而,相对于短的reads,Long Read的质量相对较低,因此目前结合Long Read进行TE多态性检测的工具较少。
近日,法国Séverine Chambeyron团队在国际著名杂志Genome Biology在线发表了“TrEMOLO: accurate transposable element allele frequency estimation using long-read sequencing data combining assembly and mapping-based approaches”的文章。
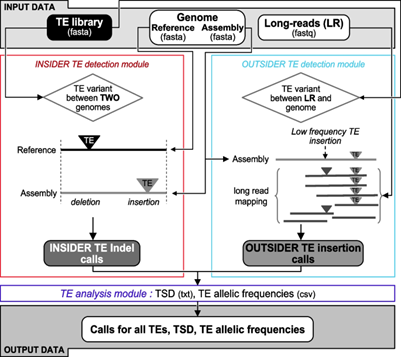
本文中,作者开发了TrEMOLO软件结合Long Read来识别TE插入和删除。其需要的输入为预先准备的TE库、参考基因组序列、单个或多个的Long Read DNA测序数据,以及用这些Long Read测序数据组装的基因组,详细参数介绍见TrEMOLO的github(https://github.com/DrosophilaGenomeEvolution/TrEMOLO)页面。
TrEMOLO作为一种新的软件,结合基于组装和比对的方法来进行转座子的检测。无论是使用高质量还是低质量的基因组组装结果,TrEMOLO都可以检测大多数的TE插入和缺失,并估计它们在群体中的等位基因频率。随后作者使用模拟数据进行了标准的Benchmark Analysis表明TrEMOLO的性能优于当前的已有工具。并将使用TrEMOLO通过使用模拟和实验数据检测到的TE插入和频率进行了验证。