本文内容速览:
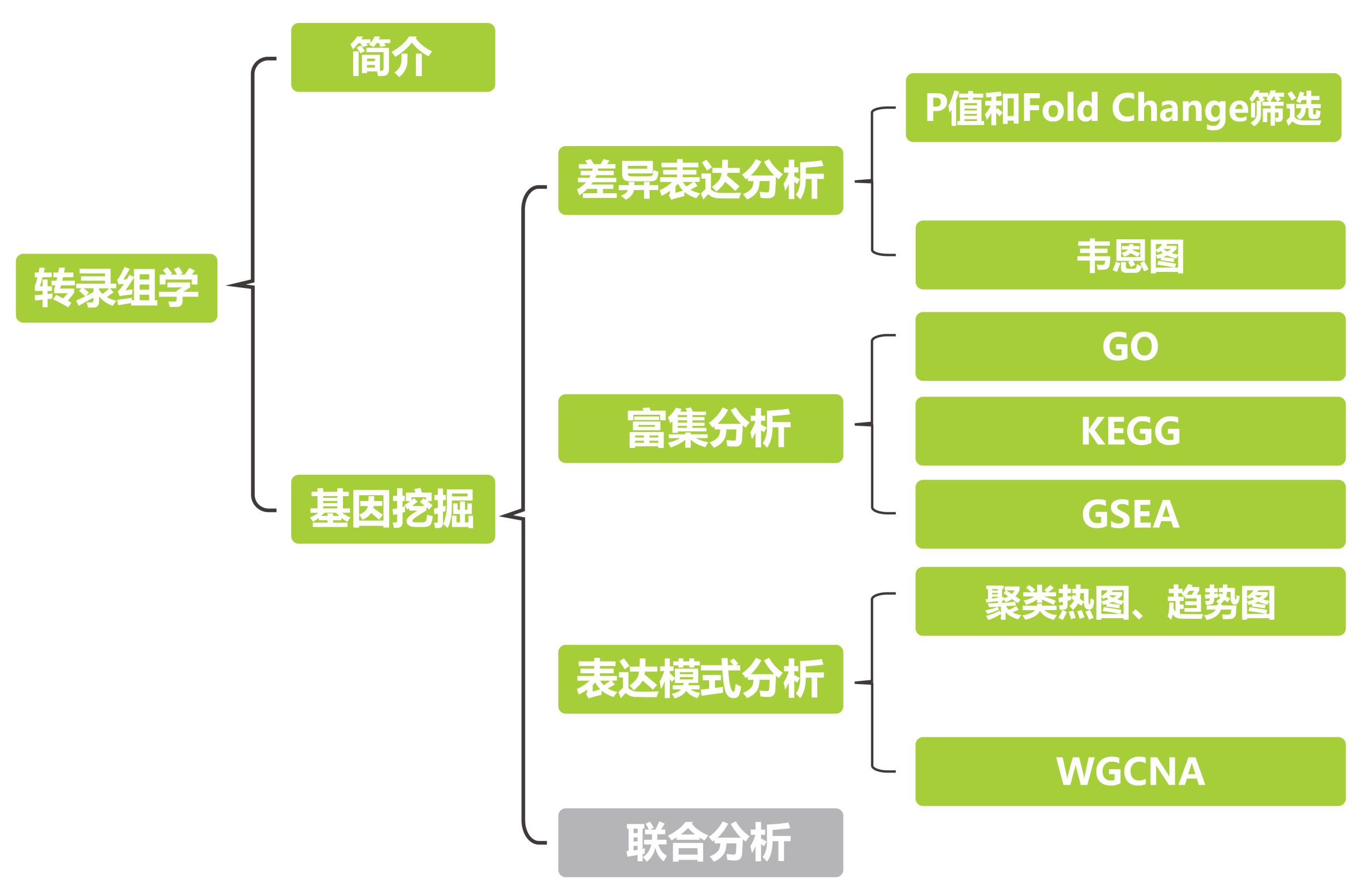
生命活动的正常执行往往依赖于机体中多层次、多功能的复杂结构系统,涉及一整套精密的表达调控机制。基因组学、表观组学、转录组学、蛋白质组学和代谢组学等多组学实验能为大家提供植物生命活动过程中多个层面的海量数据,帮助大家全面系统地挖掘在植物复杂调控网络中发挥重要功能的基因,解析从基因到表型的信息传递过程。
要理清多组学关联分析的思路,还要从理清单一组学分析的思路入手。转录组是大家目前最熟悉、使用最多的组学技术之一。在中心法则中,RNA被认为是DNA与蛋白质之间遗传信息传递的“桥梁”,同样,转录组也位于多组学的中心,可以与其他组学进行关联分析(图1)。在本次推文中伯小远就先为大家介绍利用转录组获得目的基因的方法,感兴趣的小伙伴快来看看吧!

那么什么是差异显著?统计学使用假设检验来评估差异表达分析中的显著性。主要是对两组样本基因表达量的差异倍数(Fold change)进行假设检验,并使用P值(P-value)或q值(q-value,也称为FDR)来评估假设检验中的假阳性率。假阳性率代表不是样本数据本身有差异,而是其他原因(比如取样)导致数据结果有差异,假阳性率越小,越有理由相信基因在两个样本间是有差异的。P-value表示在零假设成立的情况下,检测到的差异或更极端差异的概率,通常以P-value<0.05为显著,P-value<0.01为极显著。q-value则是P-value进行校正后的结果,它是多重假设检验后的期望值,相对于P-value更加严格。
通常将|Fold change|>2且q-value<0.05的基因称为差异表达基因(Differential expression gene, DEG)。当然这个标准也可以根据实际数据调整,保证获得一定数量范围内的上调和下调DEG,基因数量太多或太少不利于选取目的基因。如果有上万个DEG,可以选择更低的阈值,例如q-value<0.01。如果只有几十个DEG,可以将差异倍数降低,例如|Fold change|>1.5,也可以选择更宽松的P-value,例如P-value<0.05。大家可以将差异倍数高且差异极显著的基因作为候选目的基因。
差异表达分析的结果通常会以表格的形式将DEG的ID、差异倍数、显著性水平等信息展示出来,方便大家查阅和分析。但在文献中通常会用火山图、柱状图等形式更直观地呈现DEG的信息(图3、4)。

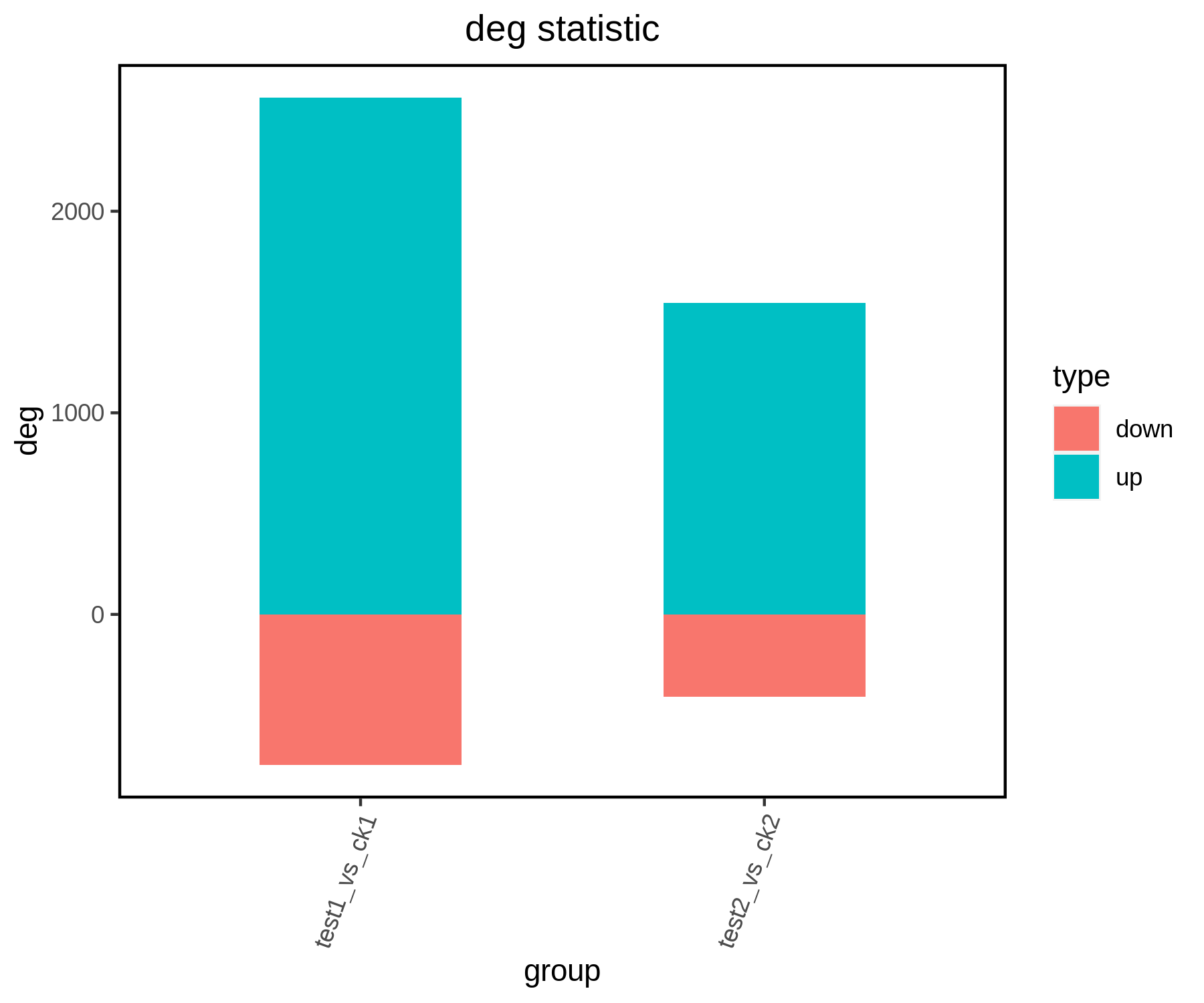
图3 DEG火山图。图中显示与样本ck1相比,样本test1中有2565个基因表达显著上调(up),748个基因表达显著下调(down)。红点表示上调DEG,绿点表示下调DEG,蓝色表示非差异表达基因(noDeg)。红箭头指出的是差异倍数高且差异极显著的基因,可以作为候选目的基因。P-value<0.05,|Fold change|>2。图片来源:伯远生物。
那什么是富集呢?富集分析涉及两个概念,前景基因和背景基因,前景基因是大家关注的基因集合(DEG集合等),背景基因就是所有基因集合。例如:在对照组和处理组两个样本的转录组测序中,前景基因就是对照组与处理组比较的DEG,背景基因就是这两个样本的所有基因。如果注释到某个生物功能的DEG占所有DEG的比例,大于注释到这个生物功能的基因占所有基因的比例,称为富集,多重检验校正表明这种富集并不是随机波动造成的,称为显著富集。举个例子,我想知道与全国相比,武汉市的大学生人数是否显著富集?在这个问题中,武汉市的人口数量就是前景基因,全国人口数量就是背景基因,“大学生”就相当于武汉市民的某个“生物功能”,还有其他的“生物功能”,例如“中学生”、“小学生”等,武汉市的大学生人数所占武汉市总人口的比例高于全国大学生在全国总人口中所占的比例,并且多重检验校正表明这种富集不是随机波动,则我们可以认为与全国相比武汉市的大学生人数显著富集。
常用的富集分析包括基因本体论(Gene Ontology, GO)富集分析和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes, KEGG)富集分析。这两者均基于统计学的超几何分布,可以将它们简单理解为先利用GO数据库或KEGG数据库对上调或下调DEG进行功能分类,再利用超几何检验计算出来P-value,根据P-value<0.05来判断是否显著,如果想要更准确一点,经过多重检验校正得到q-value,以q-value<0.05来判断DEG富集到某一生物功能或代谢途径是否显著。通过富集分析,大家可以了解差异分析得到的DEG主要具有哪些生物学功能,也可以挑选研究方向相关或显著富集的生物学功能中的基因进行功能研究。
GO是基因功能国际标准分类体系,它分为分子功能(Molecular Function, MF)、生物过程(Biological Process, BP)和细胞组成(Cellular Component, CC)三个部分,描述了基因可能行使的分子功能、所处的细胞位置、参与的生物学过程。每个基因都会对应有一个或多个GO功能条目(GO term)。GO富集分析结果会以表格的形式罗列出来,但文献中主要以柱状图、气泡图(散点图)、富集圈图和有向无环图等形式呈现(图6)。
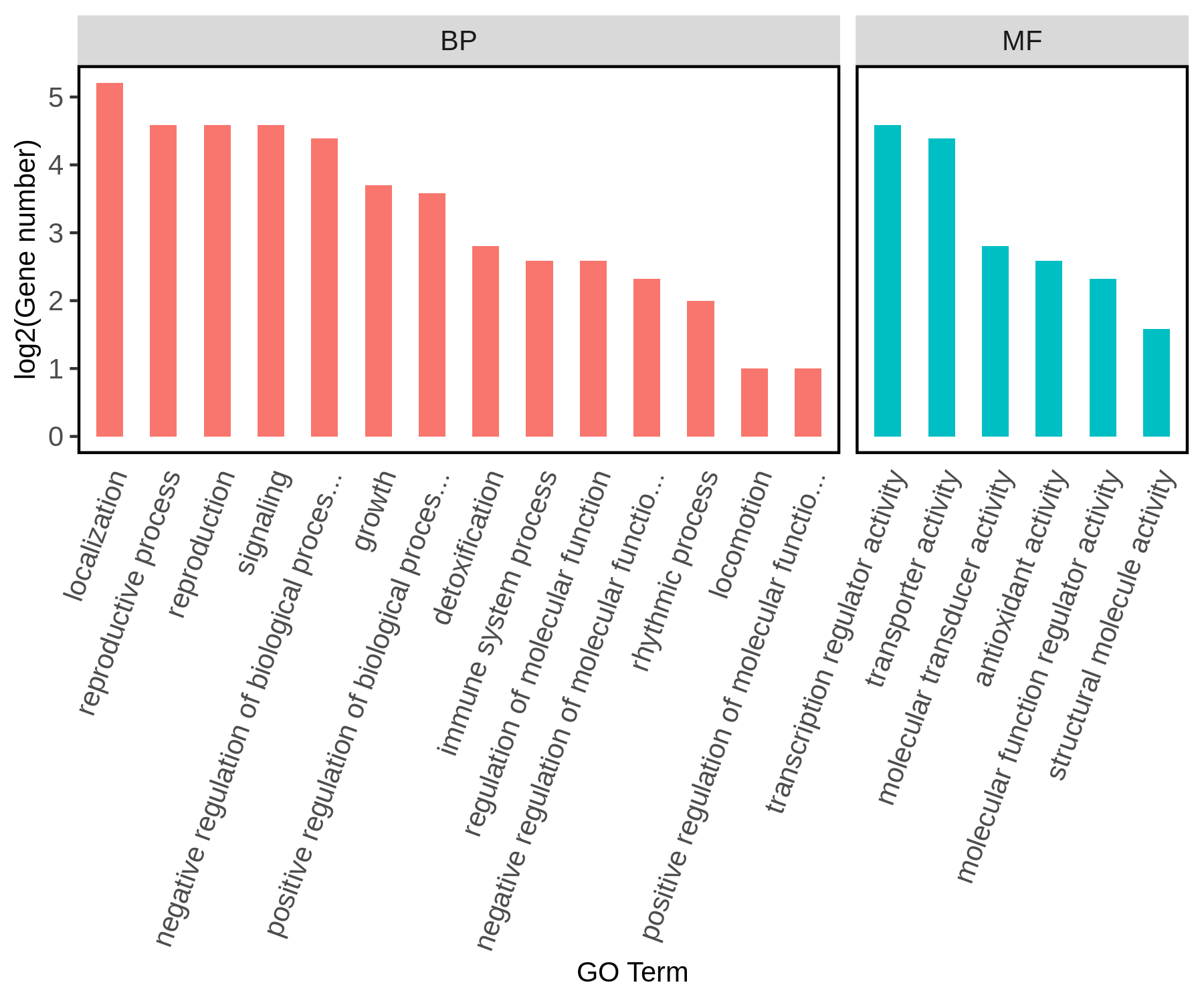
图6 GO富集分析柱状图。图中展示P-value前20的GO term,横坐标是GO term的具体名称,纵坐标是对应GO term中基因个数的log2值。图片来源:伯远生物。
KEGG是系统分析基因功能、基因组信息的数据库,KEGG提供的整合代谢途径(pathway)包括碳水化合物、核苷、氨基酸等代谢及有机物的生物降解,不仅提供了所有可能的代谢途径,而且对催化各步反应的酶进行了全面的注释,是进行生物体内代谢分析、代谢网络研究的强有力工具,有助于研究者把基因及表达信息作为一个整体网络进行研究。KEGG可视化结果包括柱状图、气泡图(散点图)、富集圈图和富集通路图等(图7)。
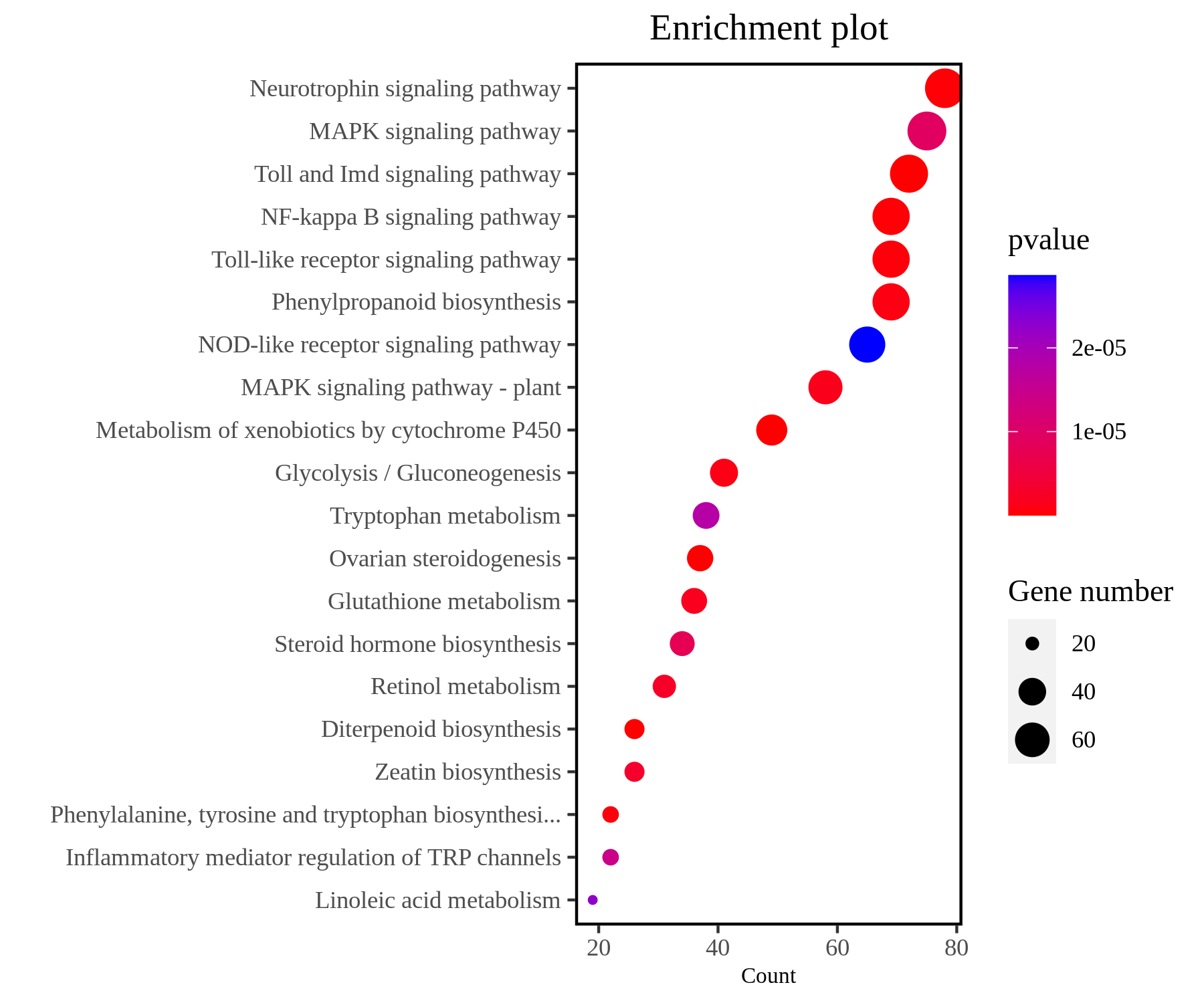
图7 KEGG富集分析气泡图。图中展示P-value前20的KEGG pathway。纵坐标是KEGG pathway的具体名称,横坐标是对应KEGG pathway中检出的基因占背景基因的个数,颜色反映P-value的大小,气泡的大小反映DEG的个数。图片来源:伯远生物。
基因集富集分析(Gene Set Enrichment Analysis, GSEA)是一种基于基因集的富集分析方法。有了GO和KEGG富集分析,为什么还要进行GSEA富集分析呢?这是因为GO和KEGG富集分析需要通过设定阈值获得显著上调或下调的DEG,容易遗漏部分差异表达不显著但有重要生物学意义的基因。同时,富集到某个通路的基因可能既有上调DEG又有下调DEG,那么这条通路总体是被抑制还是被激活是不清楚的。
GSEA的原理比较复杂,举一个简单的例子。例如:获得实验组A和对照组B的所有表达基因在KO05410这条代谢通路中富集(只是富集不一定显著)的基因集合后,GSEA会按照集合中每个基因的Fold change从大到小进行排序,接着,计算富集得分(Enrichment Score),估计富集得分的显著性水平和多重假设检验,最后会得到下面这张峰图(图8),同样也会得到P-value和q-value。峰图下面每个竖线就代表一个基因,在红色部分的基因在实验组A中表达量高,蓝色部分的基因在对照组B中表达量高。然后看某一个基因集合的所有基因主要位于这个排列顺序的红色部分还是蓝色部分,也就是峰在红色部分还是蓝色部分。如果在红色部分则表示A样本与KO05410这条代谢通路正相关,下图中显示的是正相关,说明KO05410这条代谢通路在A样本中整体被激活;如果在蓝色部分则KO05410这条代谢通路在A样本中整体被抑制。大家可以通过GSEA富集分析来判断特定生物功能的基因集合与样本或表型的关系,还可以重点关注具有显著P-value或q-value峰图中位于两侧的基因(图8中两侧竖线),例如下图峰顶左侧的基因可以认为是核心基因(Subramanian et al., 2005)。

如果样本实在太多,进行两两比较和绘制韦恩图变得繁琐且难以理清目标基因集合,而且样本是时间节点或浓度梯度这类有连续性关系的样本,可以进行表达模式分析(也称趋势分析或表达聚类分析),大家能够从所有表达基因的表达谱中快速获取我们所关注趋势的基因集合或哪种变化趋势是最显著的变化趋势,也可以获取DEG的表达模式或显著富集的代谢通路中基因的表达模式。表达模式分析的可视化结果包括聚类热图或趋势图等(图9)。

图9 DEG聚类热图。选取DEG,对这些基因在各样本中的表达情况进行表达模式分析。图片来源:伯远生物。
如果样本数大于15(建议5组以上),可以考虑开展加权基因共表达网络分析(Weighted correlation network analysis, WGCNA),WGCNA是用来描述不同样品之间基因关联模式的系统生物学方法,可以用来寻找协同表达的基因模块(module),并探索基因网络与关注的表型之间的关联关系,以及获得网络中的核心基因。从方法上来讲,WGCNA分为表达量聚类分析和表型关联两部分(图10A、B),其原理比较复杂,伯小远在这里不详细介绍了。
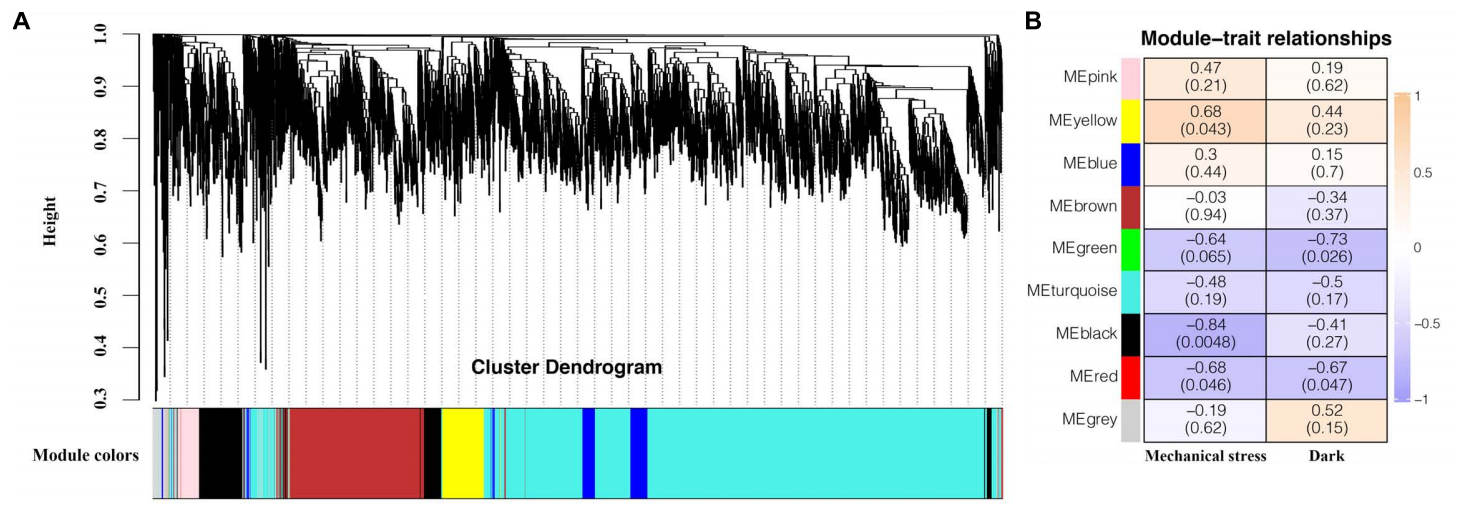
图10 WGCNA分析(Cui et al., 2019)。(A)9个模块的共表达基因的层次聚类图;(B)模块-性状特征关系图。每行表示一个模块,每列表示一个性状,方格上面的数字代表相关系数,相关系数从−1(蓝色)到1(橙色),正值代表模块与性状正相关,负值代表模块与性状负相关,括号中的数字代表显著性,其小于0.05则模块与性状相关性显著。
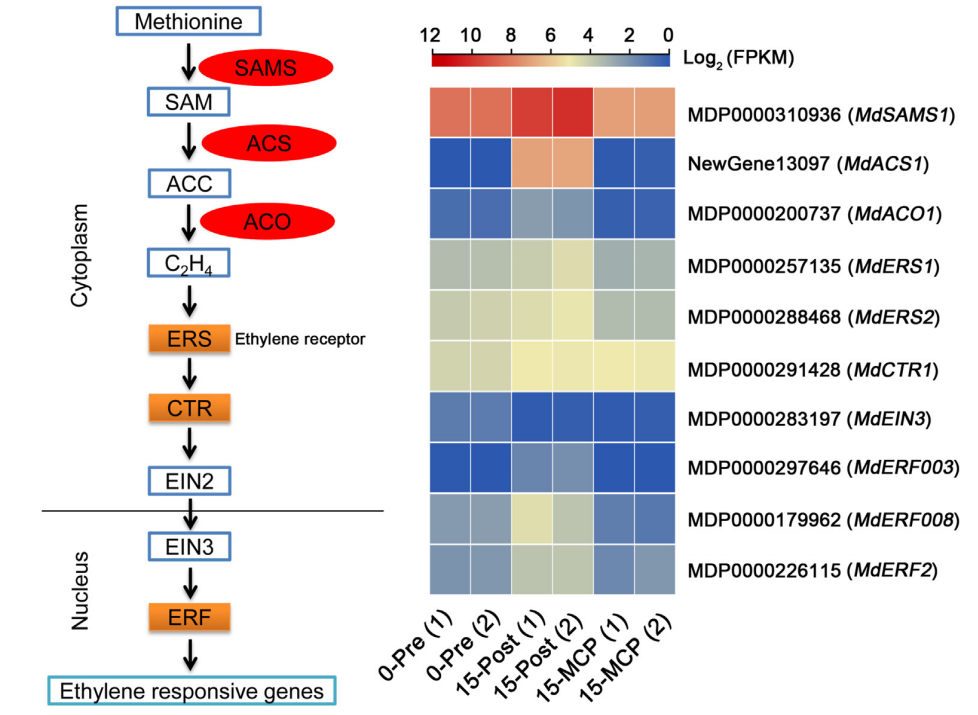
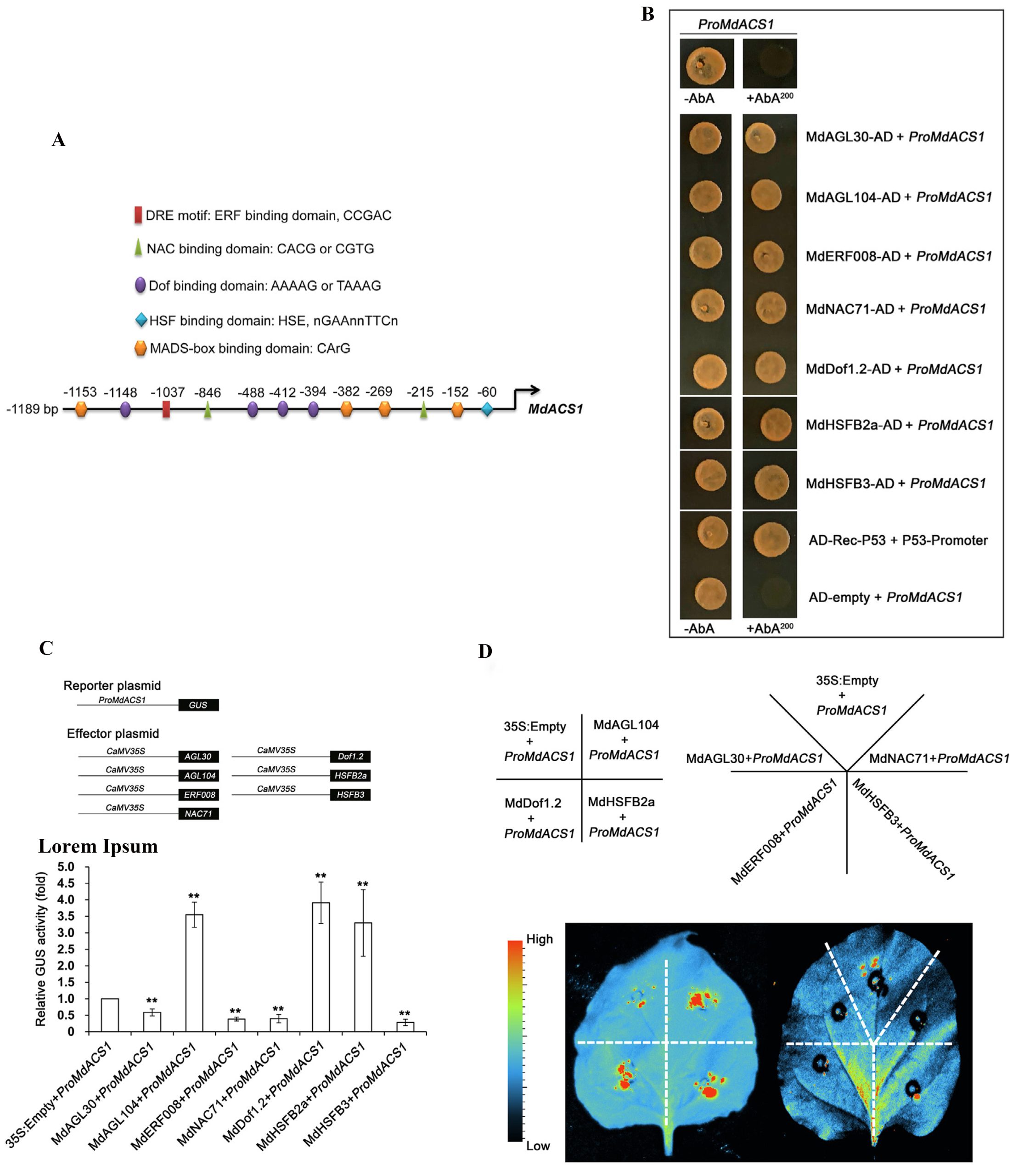
接着,作者通过酵母单杂实验(Yeast one hybrid, Y1H)和双荧光素酶报告基因实验(Dual-luciferase reporter assay, Dual-LUC)验证了这些转录因子与MdACS1的启动子结合,说明鉴定出的转录因子在果实乙烯生物合成中具有调控作用(图12)。
该研究通过共表达分析(类似于前文提到的表达模式分析)探究了参与激素代谢调节的关键转录因子,每种激素的共表达网络将基因的表达模式与激素生物合成基因相关联。以GA为例,生物活性GA由质体中的香叶酰二磷酸香叶酰(Transgeranylgeranyl diphosphate)合成,且GA的主要代谢途径已在模式植物中阐明。这些GA结构基因的表达在不同激素处理下的甘蓝型油菜中表现出不同的模式(图13a),且这些基因的转录本与306个转录因子(TF)高度相关,包括B3、bHLH、Dof和MIKC_MADS家族(图13b)。为了研究这些转录因子的潜在调控作用,作者选择了BnaDOF5.7、BnaAGL15和BnaFUS3三个转录因子进行调控网络分析,结果显示BnaDOF5.7、BnaAGL15和BnaFUS3转录本分别与7个、7个和8个GA结构基因高度相关(图13c)。
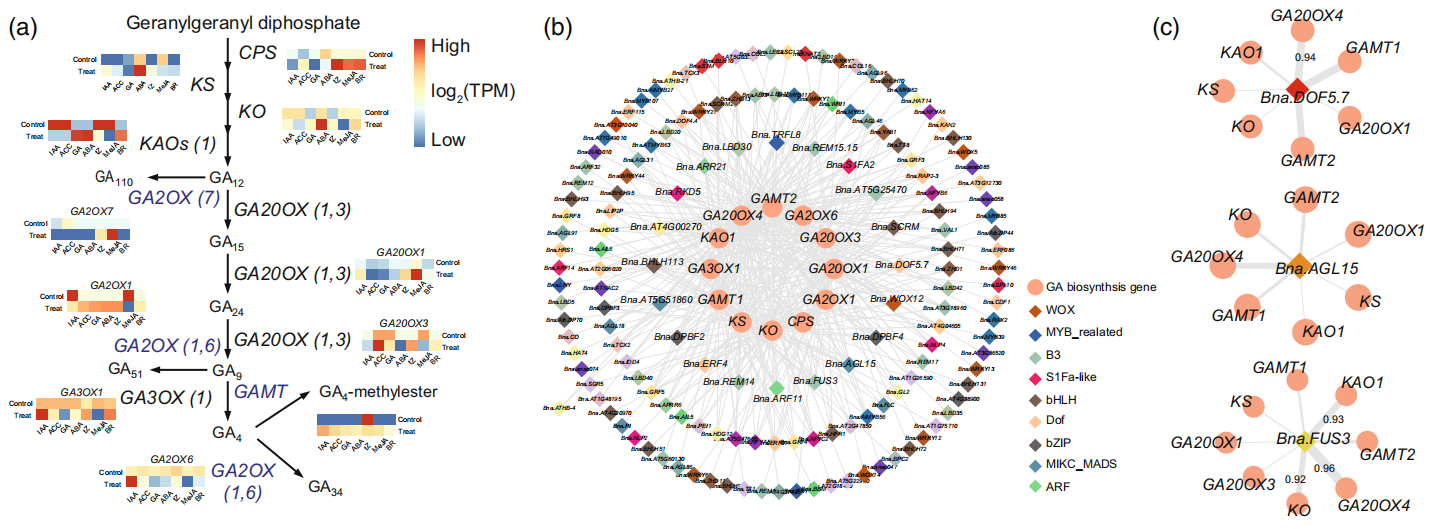
图13 寻找调控GA生物合成的关键转录因子(Liu et al., 2023)。(a)GA的生物合成途径。聚类热图表示在不同激素处理下GA生物合成途径中基因的表达模式,蓝色表示低表达量,红色表示高表达量;(b)GA生物合成的共表达调控网络。粉红色圆点代表GA生物合成基因。不同颜色的菱形点代表不同的转录因子家族,其转录物与GA生物合成基因的表达相关;(c)甘蓝型油菜GA生物合成的转录调控网络。菱形点代表调控甘蓝型油菜GA生物合成的关键转录因子。
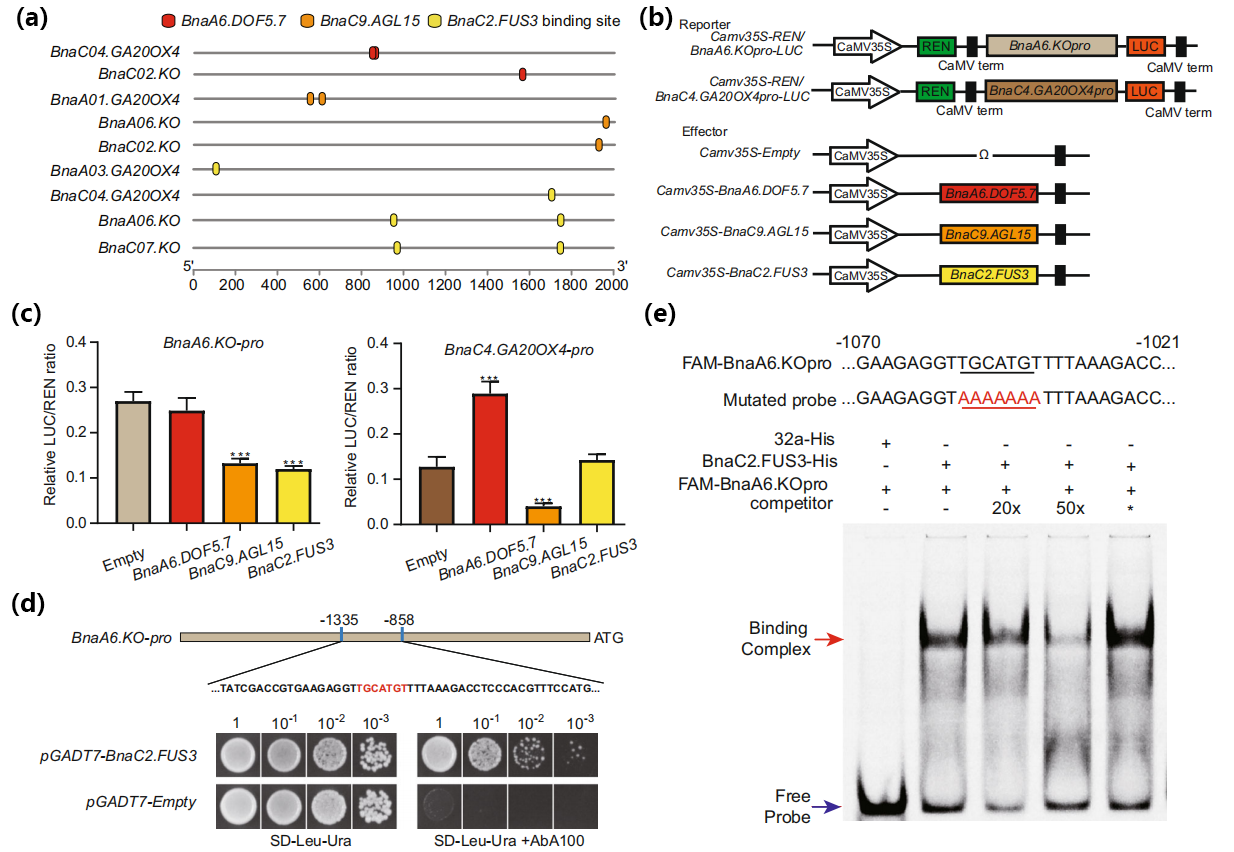
作者发现在GA生物合成相关基因的启动子序列上具有Dof、MADS和B3型TF的预测结合位点(图14a)。进一步研究BnaDOF5.7、BnaAGL15和BnaFUS3对GA生物合成相关基因的潜在调控作用,Dual-LUC实验表明BnaC9.AGL15和BnaC2.FUS3都能分别抑制BnaA6.KO的表达,BnaC9.AGL15抑制BnaC4.GA20OX4的表达,BnaA6.DOF5.7激活BnaC4.GA20OX4的表达(图14c)。BnaA06.KO启动子中有两个预测的FUS3结合位点,分别指定为P1(1049至1043bp)和P2(253至247bp)。Y1H和凝胶迁移实验(Electrophoretic mobility shift assay, EMSA)结果表明BnaC2.FUS3可以直接结合到BnaA6.KO启动子的P1位点(图14d、e)。然而,包含P2位点的区域在Y1H实验中表现出很强的自激活,EMSA实验结果显示BnaC2.FUS3和BnaA6.KO启动子的P2位点之间没有直接结合。这些结果证明BnaC2.FUS3通过特异性结合BnaA6.KO启动子的P1位点来抑制BnaA6.KO的表达。
Cui Y, Bian J, Lv Y, et al. Analysis of the transcriptional dynamics of regulatory genes during peanut pod development caused by darkness and mechanical stress[J]. Frontiers in Plant Science, 2022, 13: 904162.
