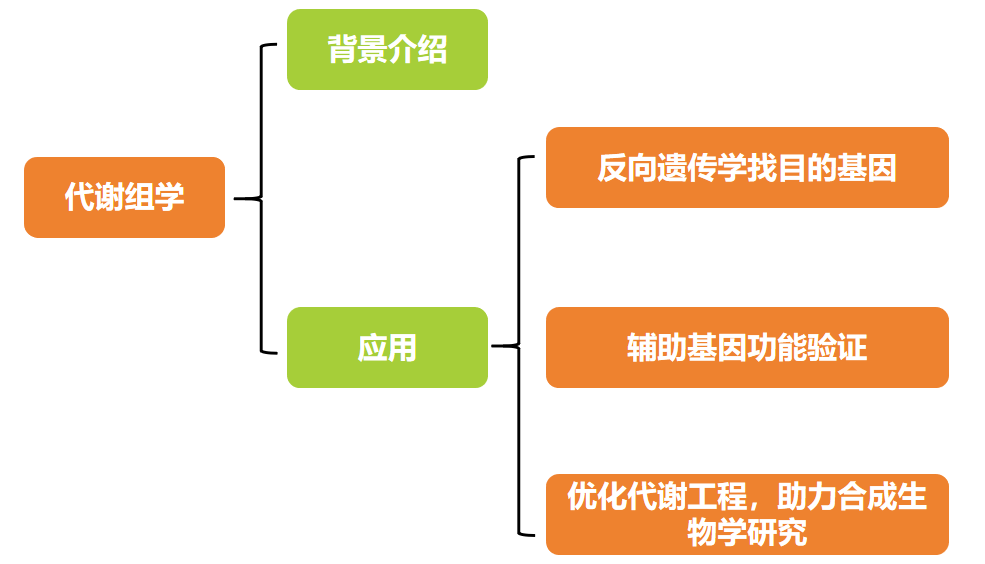
首先,作者根据内叶颜色呈黄色或白色选取了6个具有代表性的大白菜品种WWH、XQ、GCH、RWZ、XYW和RJJ为研究对象(图1A),通过代谢组学技术分析了6个大白菜品种中的总类胡萝卜素、总可溶性糖和总硫代葡萄糖苷的含量(图1B)。结果发现,黄内叶大白菜类胡萝卜素总含量和硫代葡萄糖苷总含量均显著高于白内叶大白菜,这两类物质分别决定白菜的颜色和风味。此外,黄色内叶大白菜中肌醇的含量较高(图1C),肌醇主要参与植物糖的运输和胁迫抵抗。与黄内叶大白菜相比,白内叶大白菜可溶性糖总含量显著增高,而可溶性糖的含量和种类是影响大白菜风味和甜度的重要因素。在白色和黄色内叶大白菜中共鉴定出了13种可溶性糖,其中D-果糖和葡萄糖是主要差异成分(P≤0.05),占总可溶性糖含量的96%以上。在白色和黄色内叶大白菜中共鉴定出53种差异的硫代葡萄糖苷(P≤0.05),其中脂肪型硫代葡萄糖苷占比最多约占72%,此类型的物质与大白菜的风味密切相关(图1C)。
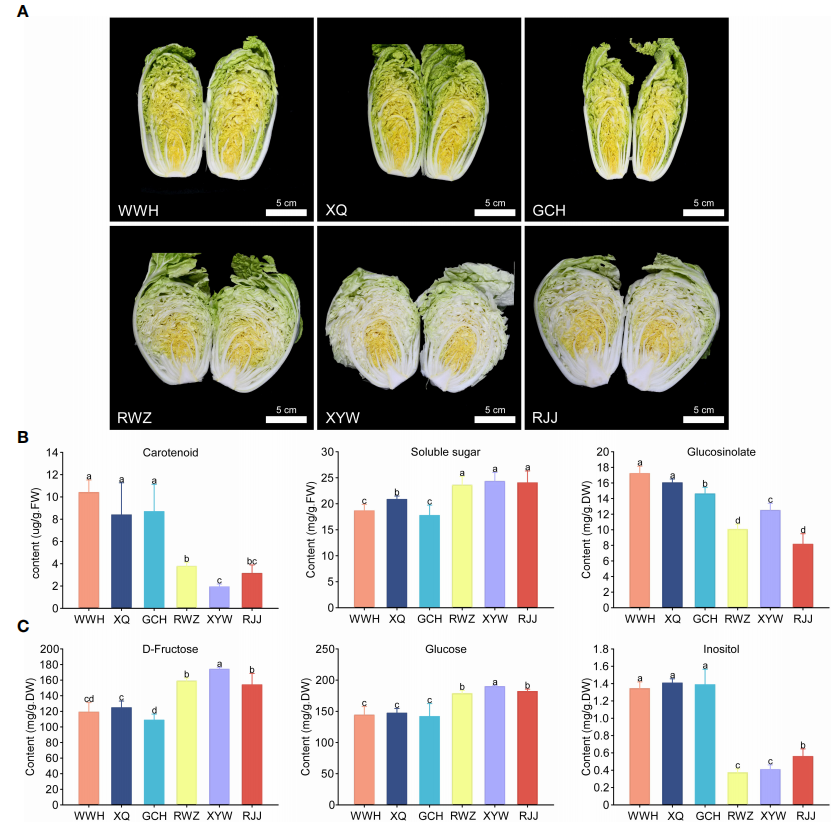
图1 黄白内叶大白菜中类胡萝卜素、可溶性糖和硫代葡萄糖苷的含量(Wang et al., 2022)。(A)黄色(WWH;XQ;GCH)和白色(RWZ;XYW;RJJ)内叶大白菜的表型。比例尺为5厘米。(B)总类胡萝卜素、总可溶性糖和总硫代葡萄糖苷含量。(C)D-果糖、葡萄糖和肌醇含量。
为了分析可溶性糖和硫代葡萄糖苷生物合成过程中可能存在的关键表达基因和调控机制,作者对6个大白菜品种进行了转录组测序(RNA-Seq)分析,根据其表达模式共鉴定出18个共表达模块(图2A)。利用“模块性状”相关性分析,发现蓝色模块中差异基因与黄内叶大白菜性状呈显著正相关,与白内叶大白菜性状呈显著负相关。蓝色模块中差异基因的KEGG富集分析(图2B)显示差异基因与大白菜的代谢密切相关,包括可溶性糖和硫代葡萄糖苷代谢。RNA-Seq的这一结果和代谢组学分析的可溶性糖和硫代葡萄糖苷含量的显著变化可相互印证。
-8.png)
图2 RNA-Seq数据结果(Wang et al., 2022)。(A)黄内叶和白内叶大白菜中共表达的基因热图。(B)蓝色模块中差异表达基因的KEGG富集分析。每个气泡代表一个代谢途径,气泡越大代表此通路富集到的基因越多,气泡颜色表示富集分析的P值。
为了绘制与可溶性糖生物合成相关的调控网络,作者对大白菜可溶性糖代谢途径进行了分析(图3A),并鉴定了与可溶性糖生物合成相关的结构基因和转录因子,利用Cytoscape软件将结构基因与转录因子进行网络分析(图3B、C)。在差异表达的基因中鉴定出6个与可溶性糖生物合成强相关的结构基因(Pearson相关系数>0.8,P<0.05),包括BraA06gSPS2、BraA03gFRK1、BraA09gFRK3、BraA01gHT4、BraA03gHT7、BraA03gHT3。根据基因功能和RNA-Seq分析,白菜中BraA01gHT4和BraA03gHT7与可溶性糖含量呈正相关,而BraA06gSPS2、BraA03gFRK1、BraA09gFRK3和BraA03gHT3与可溶性糖含量呈负相关(图3D)。根据图3A中可溶性糖代谢途径显示,HT基因主要是将果糖和葡萄糖从细胞质转运到液泡,BraA01gHT4和BraA03gHT7编码HT酶,可正向调节果糖和葡萄糖代谢,使得液泡中果糖和葡萄糖的含量增加。BraA03gHT3也编码HT酶,可负向调节果糖和葡萄糖代谢,使得液泡中果糖和葡萄糖的含量降低。另外,4个常见的转录因子BraA01gCHR11、BraA05gRAP2.2、BraA07gSCL1和BraA09gNLP6在硫代葡萄糖苷和可溶性糖的生物合成中发挥重要调控作用,转录因子的表达量与代谢物(硫代葡萄糖苷和可溶性糖)含量高度相关(图3E),表明这4个转录因子不仅参与了硫代葡萄糖苷的生物合成调控,还参与了可溶性糖的生物合成。
-9.png)
图3 大白菜可溶性糖生物合成相关途径(Wang et al., 2022)。(A)用差异表达的结构基因重建可溶性糖生物合成途径。绿色椭圆表示通过RNA-Seq鉴定到的参与可溶性糖生物合成的基因,蓝色椭圆表示通过RNA-Seq鉴定到的参与硫代葡萄糖苷生物合成的基因。(B)BraA03gHT7结构基因与转录因子的相关性网络分析。绿色圆圈代表结构基因,橙色圆圈代表转录因子。(C)BraA01gHT4结构基因与转录因子的相关性网络分析。绿色圆圈代表结构基因,橙色圆圈代表转录因子。(D)参与可溶性糖生物合成相关的结构基因。(E)参与硫代葡萄糖苷和可溶性糖生物合成的转录因子。
这篇文章主要是通过非靶向代谢组学和RNA-Seq对不同品种的大白菜进行了分析,探讨了与风味和叶色显著相关的代谢物和差异基因的表达模式。代谢组学的结果发现可溶性糖、硫代葡萄糖苷和类胡萝卜素含量显著变化。RNA-Seq发现6个与可溶性糖生物合成相关的结构基因,以及4个与可溶性糖和硫代葡萄糖苷的生物合成相关的转录因子。代谢组学的数据和转录组学的数据结果相互印证,为硫代葡萄糖苷和可溶性糖的生物合成提供了新的见解,并可解释不同叶色大白菜营养成分存在显著差异的现象。研究中找到的差异基因可以为大白菜的风味改良提供数据支持。
花粉壁的脂质成分主要由位于花药内侧的孢子绒毡层细胞合成,孢子绒毡层细胞的分化在雄性配子发育中发挥重要作用,为了弄清楚水稻花药发育过程中脂质生物合成的分子机制,作者首先从籼稻9311突变体库中分离出具有等位基因突变的雄性不育突变体sts1-1,通过回交实验发现正常可育植株和雄性不育个体的分离比约为3:1,证实sts1-1不育表型是由单个隐性基因突变引起的。
为了解STS1基因对脂质生物合成的影响,作者通过代谢组学分析了WT和突变体sts1-1花药中的脂质成分,鉴定出27类脂质共802种。与WT相比,突变体sts1-1中磷脂酰胆碱(PC)和磷脂酰乙醇(PE)(图4A)等脂类的含量明显降低,用于合成PC和PE的前体物质LPC和LPE(图4A)在突变体材料中的积累减少。作者进一步测定了WT和突变体sts1-1中脂肪酶的活性,发现突变体中脂肪酶的活性低于WT(图4B)。此外作者还发现突变体sts1-1的孢子细胞不能被脂质染料(苏丹红7B)染色(图4C)。这些结果提示STS1的基因功能障碍可能会影响水稻花药中脂质的生物合成。
图4 STS1对水稻花药中脂质生物合成的影响(Yuan et al., 2022)。(A)WT和突变体sts1-1中脂质代谢物分布情况。(B)WT和突变体sts1-1花药中脂肪酶活性分析。(C)WT和突变体sts1-1花药孢子细胞的苏丹红7B染色分析。
为了研究STS1介导脂质合成过程中是否受互作蛋白的影响,作者通过酵母双杂筛库(Y2H)实验,发现了两种介导水稻花药中脂质生物合成的蛋白,分别是OsACOS12和OsPKS2,并通过双杂点对点实验进行了验证(图5A)。此外还通过拉下实验(GST Pull-down)(图5B)和双分子荧光互补实验(BiFC)(图5C)验证了STS1分别与OsACOS12和OsPKS2相互作用。以上研究结果表明STS1可能通过与OsACOS12相互作用或与OsPKS2相互作用,调节水稻花药脂质的生物合成,影响脂质代谢,这些研究结果为水稻雄性不育育种的研究提供重要靶点。
-13.png)
图5 STS1分别与OsPKS2和OsACOS12相互作用实验结果(Yuan et al., 2022)。(A)Y2H实验。(B)GST Pull-down实验。(C、D)BiFC实验。
这篇文章作者首先从籼稻突变体库中分离出雄性不育突变体sts1-1,通过代谢组学技术发现突变体sts1-1中脂质含量显著降低,突变体sts1-1中脂质合成相关脂肪酶的活性降低。为了进一步研究STS1介导脂质合成可能存在的互作蛋白,利用蛋白互作实验证实了STS1分别与OsACOS12和OsPKS2相互作用,可能影响脂质生物合成,为阐明STS1参与水稻花药发育过程中脂质生物合成的分子机制提供重要的数据基础。
为了构建最佳的调控网络,需要对代谢调控网络中的多个靶点进行不同模式的基因编辑操作,包括增加基因表达、降低基因表达和使基因不表达(图6a)。这篇文章中作者开发了一个正交三功能CRISPR-AID系统,将转录激活(CRISPRa)、转录干扰(CRISPRi)和基因缺失(CRISPRd)整合到一个宿主中(图6b),通过质粒携带gRNAs的方式与宿主基因组融合,对代谢工程靶点进行组合优化以产生最佳表型(图6c)。
图6 CRISPR-AID系统的示意图(Lian et al., 2017)。(a)多靶点基因编辑示意图。(b)正交三功能CRISPR-AID系统。(c)CRISPR-AID系统通过探索所有可能的gRNA组合来构建最佳的调控网络。
为了评估CRISPR-AID系统经基因编辑后的效果,作者构建了一种报告酵母菌株,对于CRISPRa,mCherry由中等强度的CYC1p启动子驱动;对于CRISPRi,mVenus由强启动子TEF1p驱动;对于CRISPRd,ADE2基因缺失会导致在缺乏腺嘌呤合成的培养基中形成红色菌落。选取dSpCas9-VPR、dSpCas9-MXI1和SpCas9作为阳性对照。结果显示在报告酵母菌中mCherry基因的表达增加了5倍以上(图7a),mVenus基因的表达降低了10倍左右(图7b),ADE2基因约95%以上不表达(图7c)。
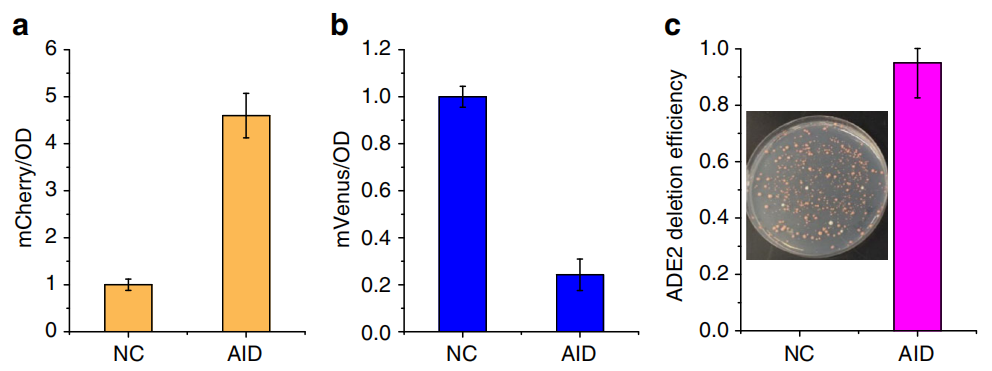
图7 利用报告酵母菌展示CRISPR-AID系统的调控效果(Lian et al., 2017)。通过用含有3个gRNAs的单质粒转化报告酵母菌株,同时实现了mCherry(a)的转录激活、mVenus(b)的转录干扰和内源基因ADE2(c)的不表达。
接下来作者通过测定酵母中β-胡萝卜素的含量来评估CRISPR-AID系统是否可以稳定的应用于代谢工程的研究。前期研究发现,HMG1基因是编码甲羟丙酸途径的主要限速酶,ERG9基因与β-胡萝卜素生物合成和内源性甾醇生物合成相关,ROX1基因编码应激响应相关的转录调控因子,HMG1基因表达上调、ERG9基因表达下调以及ROX1基因不表达可显著增加β-胡萝卜素的含量。因此作者选取HMG1、ERG9和ROX1分别做为CRISPRa、CRISPRi和CRISPRd的三个靶点(图8a)。通过RT-qPCR技术证实HMG1基因表达增强(图8c)、ERG9基因表达下降(图8c)以及ROX1基因基本不表达(图8d)。HMG1基因表达增强使得β-胡萝卜素含量增加1.5倍,这可能是甲羟戊酸途径的总代谢通量增加所致。ERG9基因表达下降使得β-胡萝卜素的含量降低至原含量的3/10(图8b)。而当HMG1基因表达增强和ERG9基因表达下降相结合时,β-胡萝卜素的含量提升了2.5倍(图8b)。当HMG1基因表达增强、ERG9基因表达下降以及ROX1基因基本不表达时,β-胡萝卜素的含量提升了约3倍(图8b)。
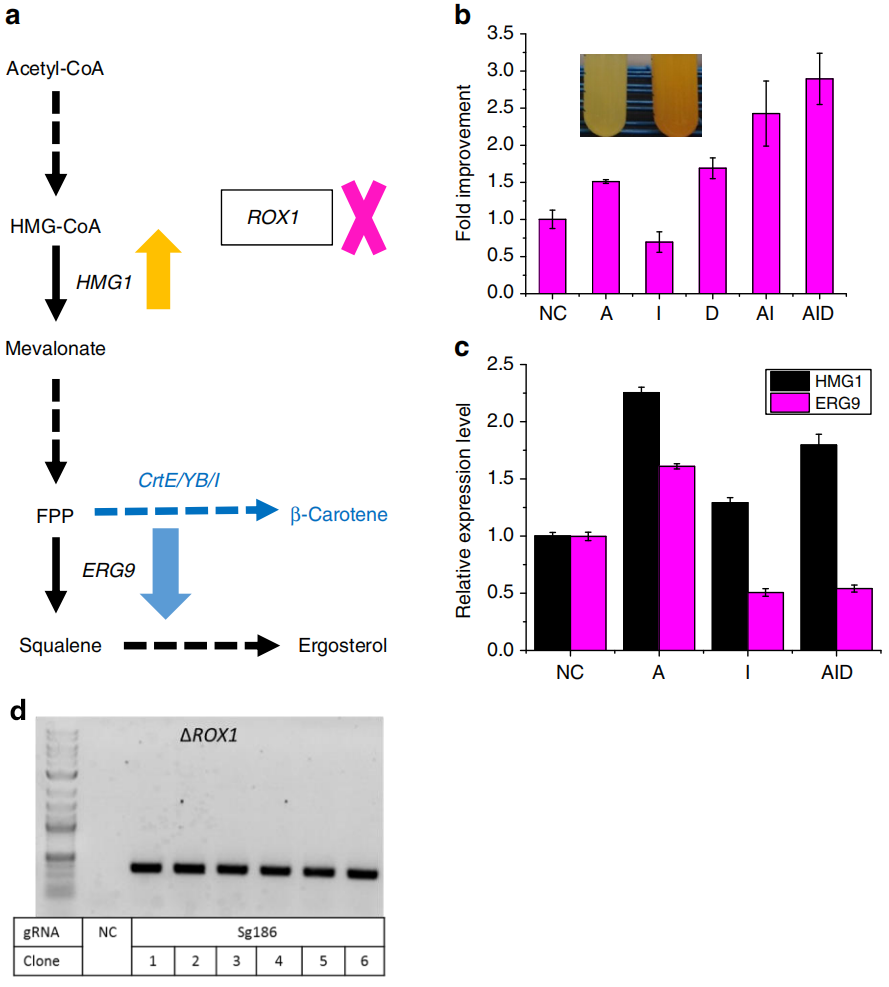
图8 用于代谢工程的CRISPR-AID系统(Lian et al., 2017)。(a)β-胡萝卜素的生物合成。选择HMG1、ERG9和ROX1分别作为CRISPRa、CRISPRi和CRISPRd的三个研究靶点。(b)单gRNA编辑(A-pSg175、I-pSg172和D-pSg186)、双gRNA编辑(AI-pSg585)和三gRNA编辑(AID-pSg239)对β-胡萝卜素含量的影响。利用RT-qPCR验证CRISPRa(HMG1,图c)、CRISPRi(ERG9,图c)和CRISPRd(ROX1,图d)表达水平。
这篇文章中作者开发了一个CRISPR-AID系统,通过多靶点协同的方式对代谢工程的靶点选择进行了优化,通过报告酵母菌评估了CRISPR-AID系统基因编辑的效率,利用代谢组学技术分析CRISPR-AID系统对β-胡萝卜素含量的影响,最终对β-胡萝卜素代谢网络进行优化。基于代谢组学技术可以找到最优的代谢途径和关键的调控酶,为代谢工程的改进提供重要参考,也为合成生物学的快速发展提供技术保障。
References:
Patel M K, Pandey S, Kumar M, et al. Plants metabolome study: Emerging tools and techniques[J]. Plants, 2021, 10(11): 2409.
Anzano A, Bonanomi G, Mazzoleni S, et al. Plant metabolomics in biotic and abiotic stress: a critical overview[J]. Phytochemistry Reviews, 2021: 1-22.
Wang L, Zhang S, Li J, et al. Identification of key genes controlling soluble sugar and glucosinolate biosynthesis in Chinese cabbage by integrating metabolome and genome-wide transcriptome analysis[J]. Frontiers in Plant Science, 2022, 13: 1043489.
Yuan G, Zou T, He Z, et al. SWOLLEN TAPETUM AND STERILITY 1 is required for tapetum degeneration and pollen wall formation in rice[J]. Plant physiology, 2022, 190(1): 352-370.
Lian J, HamediRad M, Hu S, et al. Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system[J]. Nature communications, 2017, 8(1): 1688.
文章来源:奇点网
