文章整体思路:
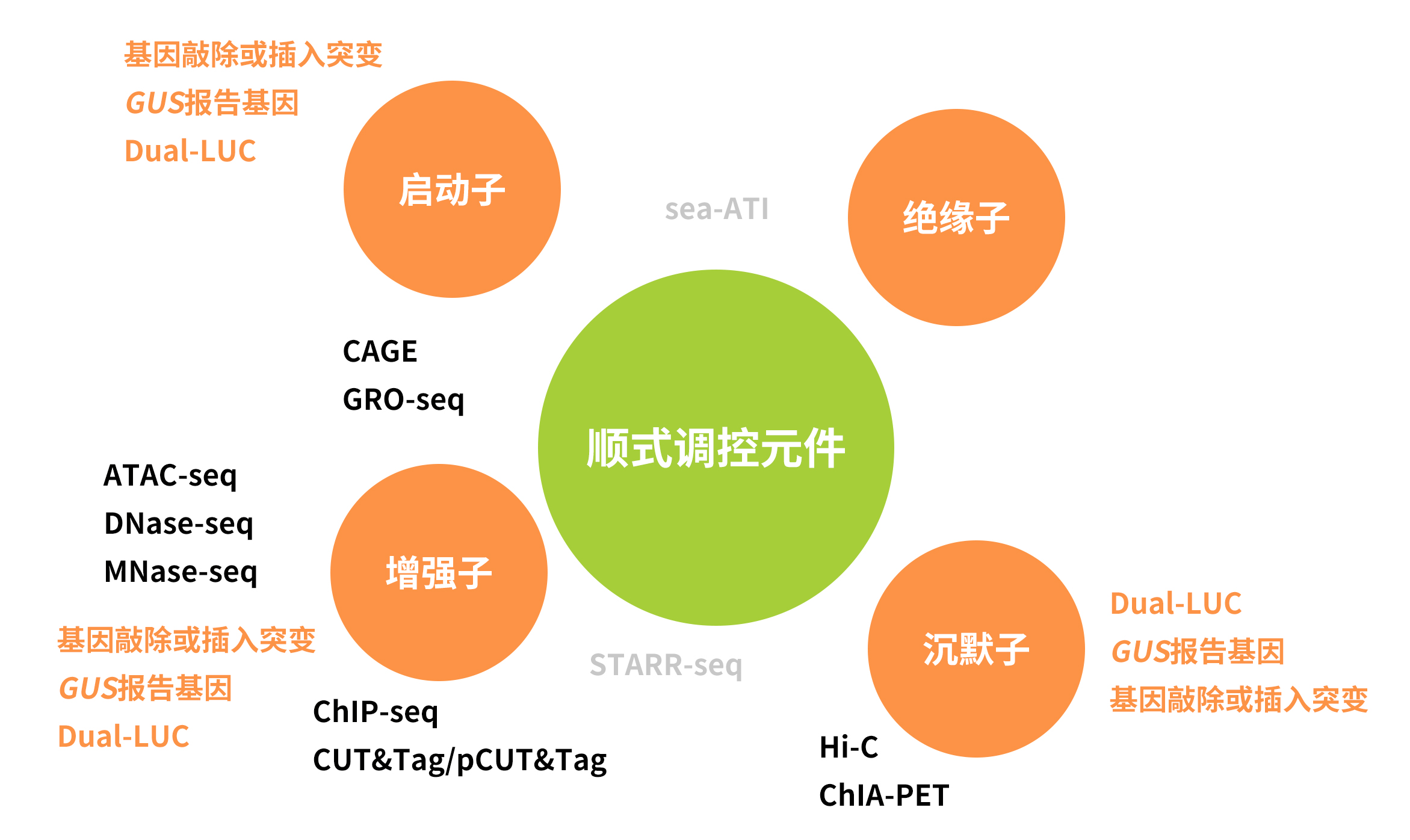
基因的表达模式是由顺式调控元件(Cis-regulatory elements,CREs)介导的,这些元件上有转录因子(Transcription factors,TFs)识别并结合的位点。CREs通常聚集在一起形成顺式调节模块(Cis-regulatory modules,CRMs)(Marand et al., 2023)。越来越多的研究表明,CREs能够确保基因在特定的时空模式下进行表达,这对动植物维持正常的生长发育过程和在环境胁迫下做出反应是必需的。下面,伯小远就为大家介绍一下这些调控基因表达的“幕后工作者们”,以及它们的研究方法。
转录区域发生修饰也可以激活转录或抑制转录。 例如,+1核小体(转录起始位点后的第一个核小体)中的H2A.Z发生单泛素化修饰在转录抑制中起着至关重要的作用,H2A.Z发生乙酰化修饰在转录激活中起着关键作用(Gómez-Zambrano et al., 2019)。如果你想了解关于启动子的更多知识可以阅读往期文章“【干货】启动子研究方法高质量总结”。

(2)寻找目的基因的下游调控启动子:
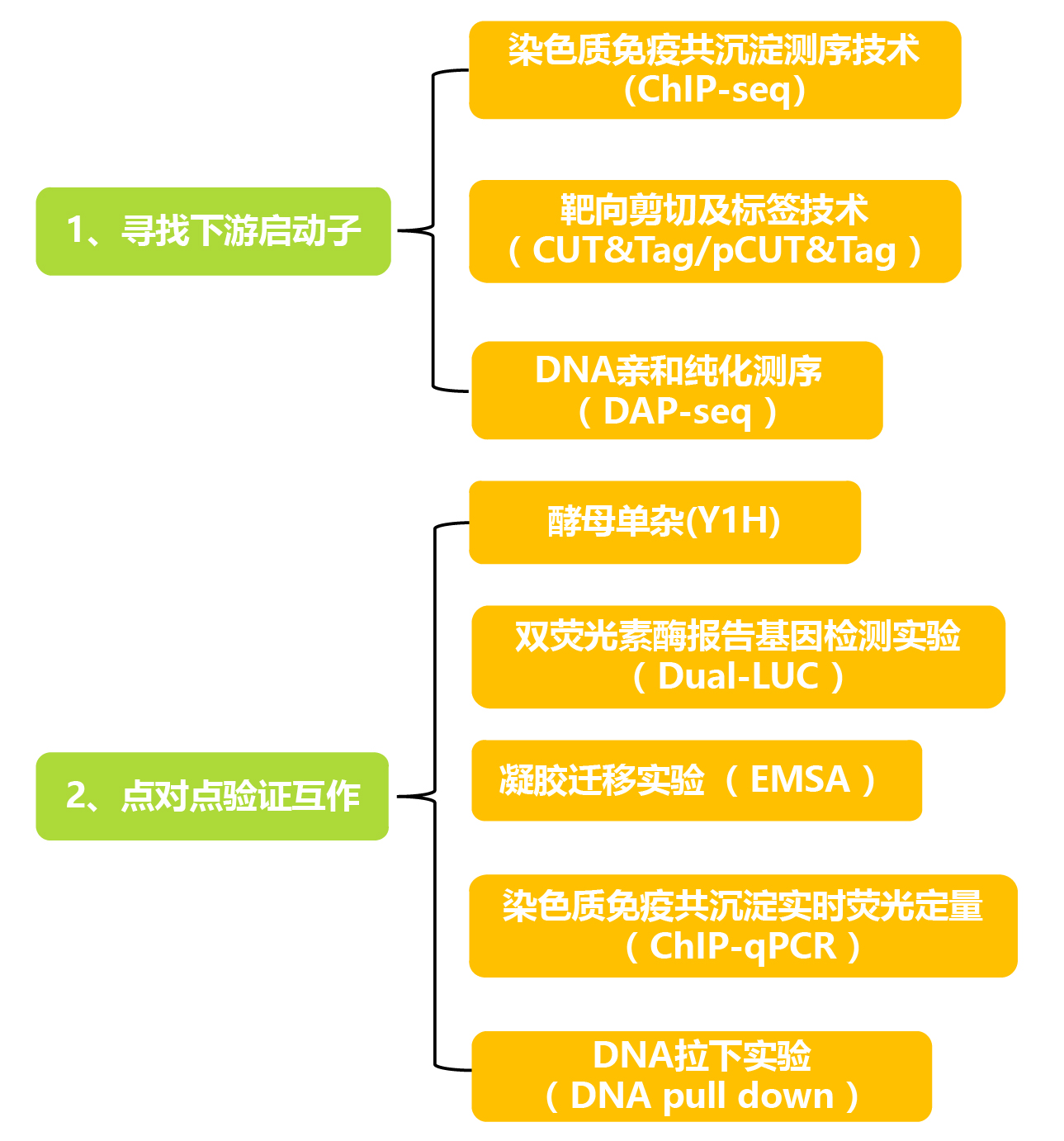
一般根据实验材料、适用范围以及优缺点选择合适的表观组学技术来寻找下游调控基因,这些表观组学技术的区别如下:
表1 寻找下游调控基因的表观组学技术的比较。

验证目的蛋白和下游调控基因的启动子序列相互作用时,一般需3种或3种以上的方法同时进行证明,遵循体内实验与体外实验相结合的原则。点对点验证实验方法的优缺点如下:
表2 验证目的蛋白和启动子序列相互作用的实验方法的比较。

科学家们开发了许多种鉴定方法,如CAGE、RAMPAGE、STRT等。2018年,美国剑桥麻省理工学院Joshua Levin教授课题组在Nature Methods上发表了题为“Comprehensive comparative analysis of 5’-end RNA-sequencing methods”的文章,该文章比较了六种鉴定TSS的5’端RNA-seq方法,发现CAGE(Cap analysis of gene expression)技术表现最优(Adiconis et al., 2018)。

CAGE是一种鉴定TSS和基因表达水平检测的技术。CAGE技术的主要原理是对mRNA 5’端“帽子”加上生物素(图1),利用链霉亲和素对生物素进行捕获,从而捕获带有“帽子”的mRNA,然后将其反转录出5’端完整的cDNA,再通过对cDNA的高通量测序,在全基因组范围内检测出TSS和基因转录情况。CAGE技术能够作为微阵列芯片和RNA-seq的替代品(Adiconis et al., 2018)。
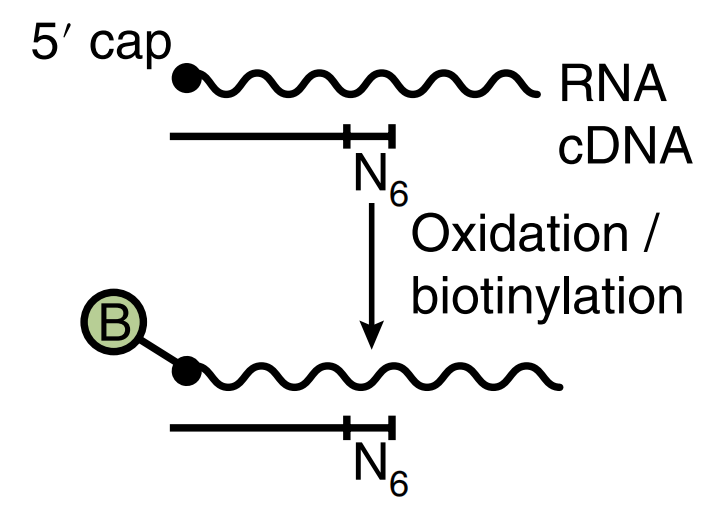
图1 CAGE-seq技术的原理(Adiconis et al., 2018)。
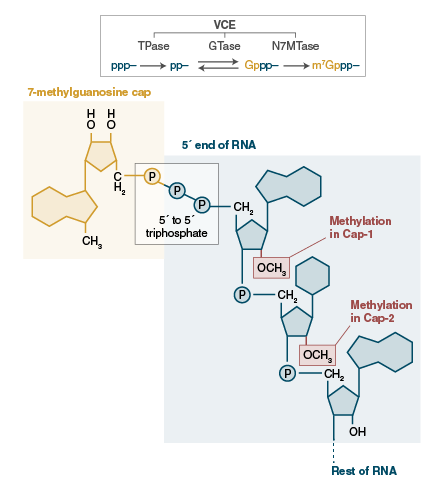
这里的“帽子”是什么?这里我们回顾一下分子生物学知识,前体mRNA需要经过内含子剪切和外显子的拼接、5’端加“帽子”(7-甲基鸟苷)、3’端加“尾巴”(poly A),以及碱基上的化学修饰等等,才能变为成熟的mRNA。TSS是转录开始的第1个核苷酸,是加帽位点,加帽能保护mRNA的5’端不被磷酸酶和核酸酶破坏,并能促进翻译的起始。

在植物中,对增强子的研究刚刚兴起。有研究表明,植物增强子的序列特征与多细胞动物中定义的增强子特征既有共同点也有不同点。例如,组蛋白H3K4me1是在人体中普遍存在的一种活性增强子标记,但在拟南芥的远端增强子中并没有发现(Yan et al., 2019);而人体中的另一种活性增强子标记,组蛋白H3K27ac,在玉米的远端增强子中被鉴定出来(Oka et al., 2017)。目前的研究结果表明,植物活性增强子不仅与H3和H4乙酰化有关,如H3K9ac、H3K27ac、H4K12ac,还与H3K4me3、H3K36me3有关,而非活性增强子与H3K27me3有关。
在动物中,增强子转录本(eRNA)的存在已被证明是活性增强子的标志, 在细胞中eRNA的丰度很低,同时容易被降解,需要比mRNA更灵敏的检测方法,如GRO-seq(Genome-wide nuclear run-on assays)技术。活性增强子通常位于可及性的染色质区域,该区域的基因组序列对核酸酶活性敏感,也称为DNase I超敏位点(DNase I hypersensitive sites,DHSs),DNase-seq技术就是利用DNase I来检测DHS的方法。
增强子还可以嵌入它们所调控的基因中,在植物中有研究表明,内含子中会含有增强子元件,例如,拟南芥中的AGAMOUS(AG)(Sieburth et al., 1997)和玉米中的knotted1(kn1)(Inada et al., 2003)。大家如果想要了解更多与增强子相关的内容可以阅读往期文章“如何研究“暗物质”——增强子?”。

在之前的研究中,通过检测DHS能够成功地预测位于拟南芥基因间区域的增强子。于是,研究者对拟南芥全基因范围内的内含子型DHS进行检测,在幼苗组织中鉴定出与806个基因相关的941个内含子型DHS,在花组织中鉴定出与1069个基因相关的1275个内含子型DHS(图4A)。
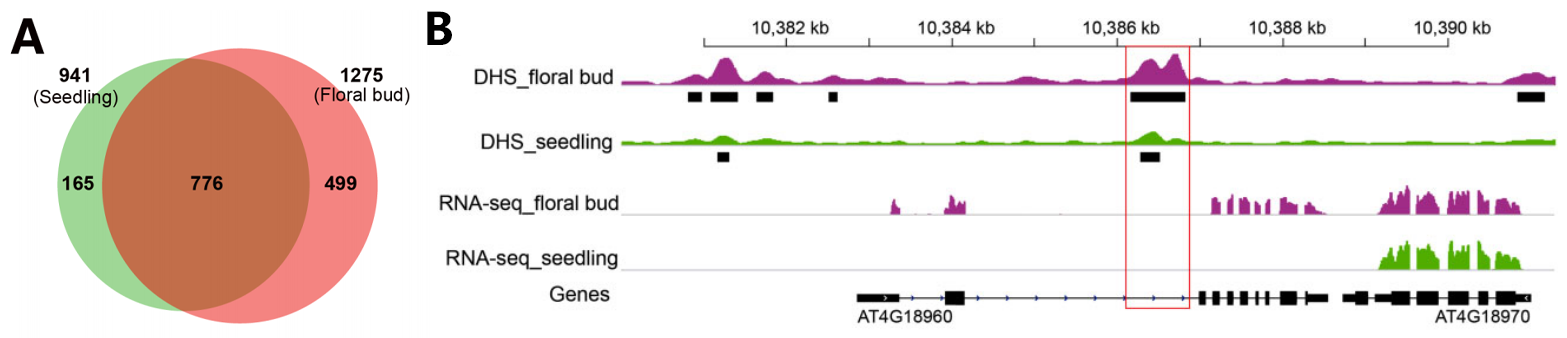
图4 对拟南芥内含子型DHSs的分析(Meng et al., 2021)。(A)在幼苗组织和花组织中鉴定的内含子DHSs的维恩图(Venn diagram);(B)在AGAMOUS基因(AG;AT4G18960)的第二内含子中发现了DHS。该内含子DHS调节AG表达的功能在先前的研究中已经得到证实,具有增强子功能。黑色长框代表DHS区域。峰高度表示DNase-seq读取reads的富集程度。红框中是包括DHS的内含子区域。
研究者使用了一种基于GUS报告基因的系统来对21个内含子型DHS进行功能验证。这种GUS报告基因系统是指将DHS序列融合mini 35S启动子和GUS报告基因的载体(图5A),并使用仅具有mini 35S启动子和全长35S启动基因的载体分别用作阴性和阳性对照,进行遗传转化获得转基因株系。每个DHS载体获得了14–31个独立的转基因株系,在每个转基因株系的不同发育阶段进行GUS测定,只有当超过50%的转基因株系在相同组织中显示出一致的GUS表达信号时,DHS才被认为具有增强子功能。实验结果显示,预测的21个内含子DHS中有15个(71%)被证实为具有增强子功能。
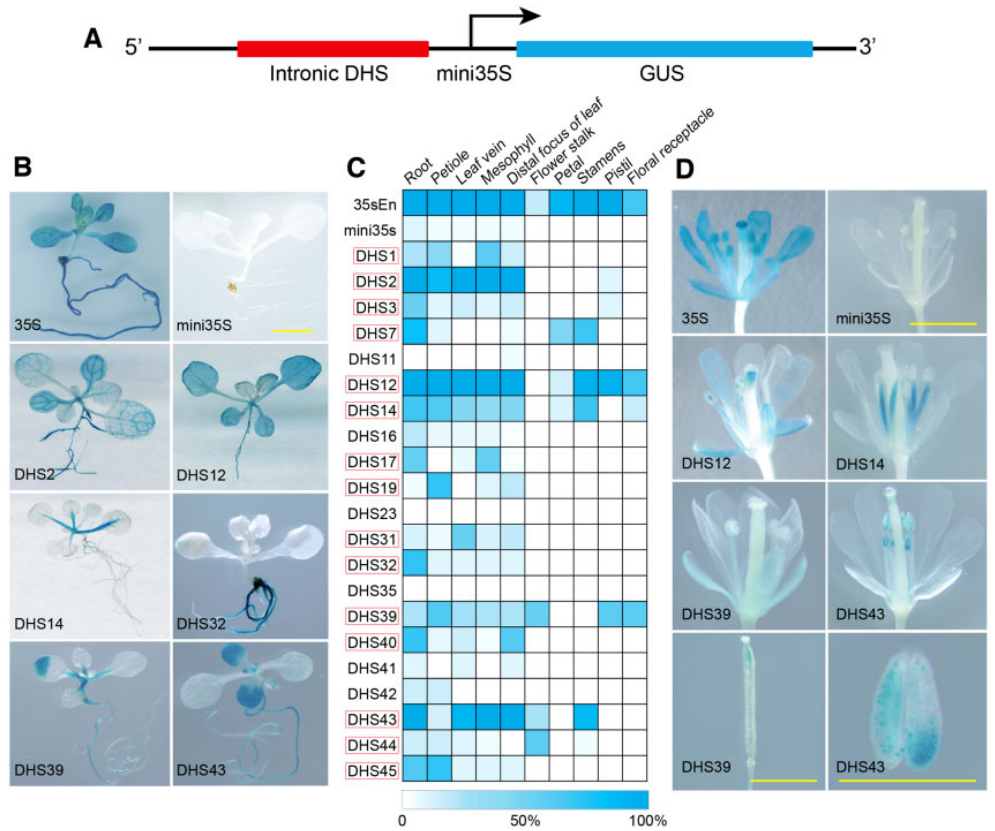
图5 基于GUS报告基因系统对内含子型DHS进行功能验证(Meng et al., 2021)。(A)用于验证增强子功能的GUS融合载体的结构示意图;(B)在叶片和根中显示GUS信号的部分转基因幼苗。DHS的名称标记在图片上。35S代表全长35S启动子;(C)不同转基因株系在不同组织中的GUS表达情况的热图。白色表示在该转基因株系的所有植株中均未检测到GUS的表达。深蓝表示在该转基因株系的所有植株中都检测到GUS表达。红框标记了被验证具有增强子功能的15个DHS;(D)处于开花阶段的代表性转基因株系在花的不同部位显示的GUS表达信号。
DHS7位于MYB相关转录因子TRIPTYCHON(TRY)基因的第二个内含子中,TRY促进根毛的形成,抑制叶苗的形成,并参与调控拟南芥开花过程。高分辨率的微球菌核酶测序(MNase-seq)显示,DHS7是由DHS7#1和DHS7#2组成的复合型增强子。GUS报告基因系统的结果显示,DHS7#1只在花中激活GUS的表达,而DHS7#2在叶片和根中激活GUS的表达。
为了进一步验证DHS7#1和DHS7#2增强子的功能,研究者利用CRISPR/Cas9对DHS7#1和DHS7#2增强子分别进行单敲除以及联合双敲除。敲除增强子DHS7#1导致21d叶片中TRY表达降低,敲除株系表现出晚花性状;敲除增强子DHS7#2导致21d叶片中TRY表达升高,敲除株系莲座叶的叶毛分支增加和成簇生长;而DHS7#1和DHS7#2双敲株系(166~187bp)既表现晚花又表现叶毛分支增加和成簇生长(图6)。
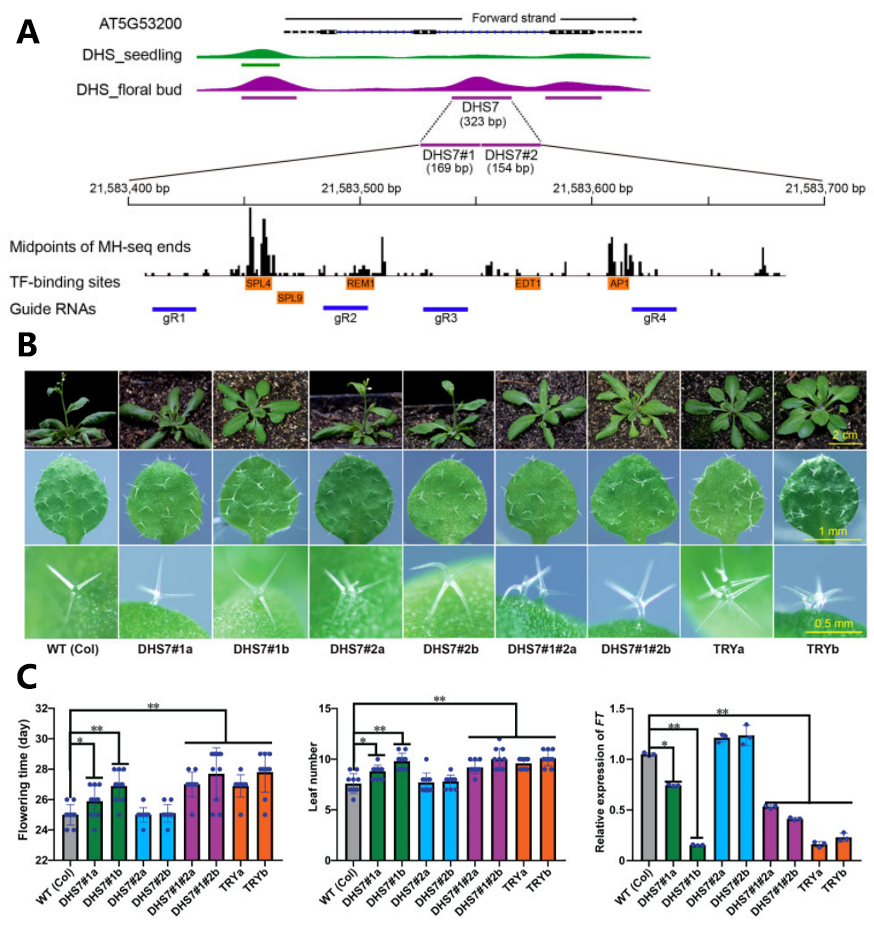
图6 DHS7增强子的序列情况以及DHS7增强子序列的敲除表型(Meng et al., 2021)。(A)TRY(AT5G53200)基因序列及其相关DHS的可视化图谱。DHS7(323bp)在花组织中被鉴定出来,并且位于第二内含子中。DHS7区域扩展显示出DHS7#1(169bp)和DHS7#2(154bp)的序列位置。图中展示了DHS7中含有可能与五个转录因子(TFs)PL9、SPL4、REM1、EDT1和AP1结合的motif,以及sgRNA在基因组上的位置;(B)野生型(WT)、DHS7#1和DHS7#2单/双敲突变体以及TRY缺失突变体的开花时间和莲座叶的叶毛表型;(C)野生型(WT)、DHS7#1和DHS7#2单/双敲突变体以及TRY缺失突变体的开花时间,叶的数量以及TRY在21天龄叶组织中的表达情况。
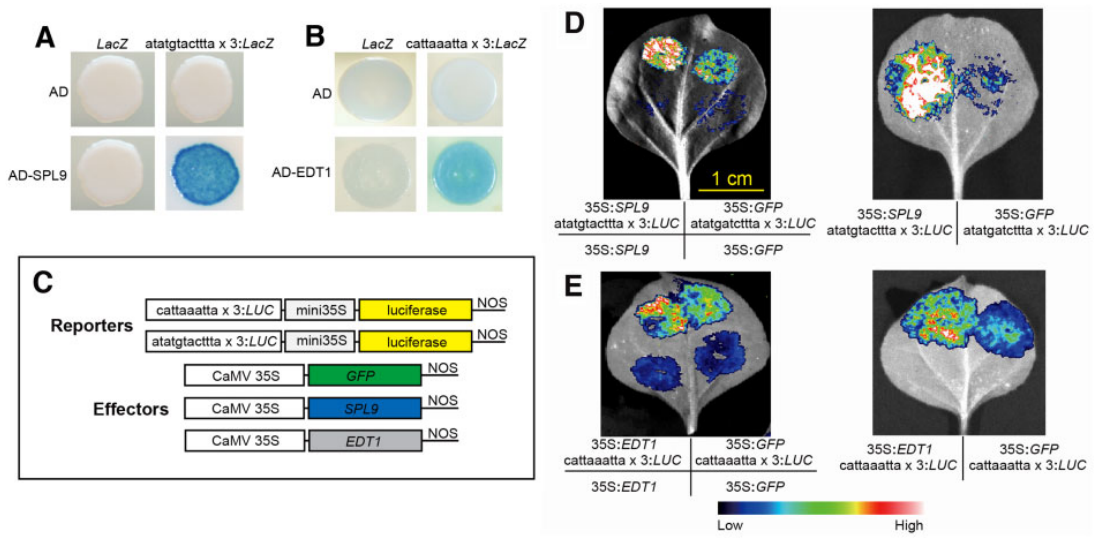
酵母单杂实验(Y1H)和双荧光素酶报告基因检测实验(Dual-LUC)验证了SPL9能够与DHS7#1增强子中“atatgtacttta”序列结合,EDT1能够与DHS7#2增强子中“cattaaatta”序列结合(图7)。
(2)将预测增强子序列融合mini35S和GUS报告基因可以鉴定增强子的功能,类似于启动子活性分析的方法。
(3)可以利用Y1H和Dual-LUC来验证增强子与转录因子预测的结合位点。

表3 研究染色质可及性方法的比较。

表4 研究组蛋白修饰方法的比较。
2023年,华中农业大学李兴旺教授和李国亮教授带领的作物三维基因组学创新团队在Cell Reports发表了题为“Haplotype mapping of H3K27me3-associated chromatin interactions defines topological regulation of gene silencing in rice”的研究论文。该研究首次在水稻基因组中系统鉴定了类沉默子和类超级沉默子调控元件(Ouyang et al., 2023 )。我们以这篇文章为例,来看看寻找沉默子和鉴定沉默子功能方法。

通过利用ChIP-seq技术检测H3K27me3位点,研究者首次在水稻基因组中系统寻找到了类沉默子(typical silencer-like regions,TSRs)和类超级沉默子(Super silencer-like regions,SSRs)调控元件。研究者发现H3K27me3关联基因的表达水平显著地低于非H3K27me3关联的基因,并且SSR关联的基因明显低于TSR关联的基因的表达水平(图8C),表明预测的沉默子元件具有基因转录抑制功能。
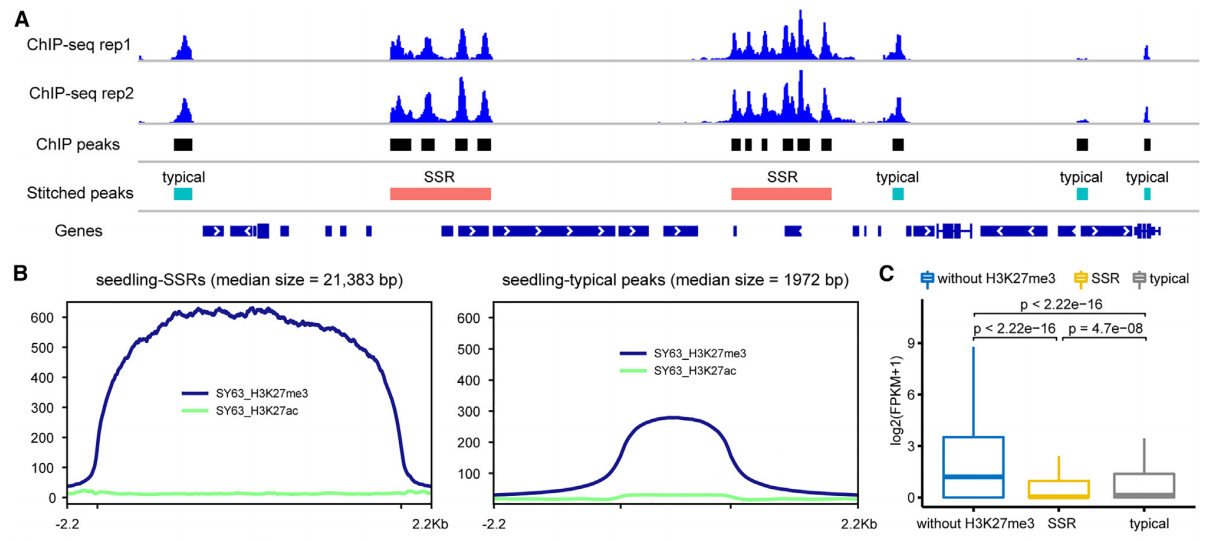
图8 全基因组范围内鉴定SSR和TSR(Ouyang et al., 2023 )。(A)代表性SSR和TSR的基因组图谱;(B)SSR和TSR信号峰宽度和强度的比较;(C)非H3K27me3关联基因、SSR关联基因、TSR关联基因表达水平的比较。
为了进一步验证类沉默子的转录抑制功能,研究者对18个类沉默子进行了双荧光素酶报告基因实验(Dual-LUC),具体是将这些元件构建到双荧光素酶报告载体pMY155中,然后将重组载体瞬时转染本氏烟草叶,仅携带mini 35S启动子的pMY155载体为阴性对照(图9A)。实验结果显示,18个类沉默子中有13个(72%)与35S启动子的对照相比,荧光素酶信号较低,表现出转录抑制活性(图9B、C)。这些结果进一步证实了H3K27me3关联的类沉默子的转录抑制功能。
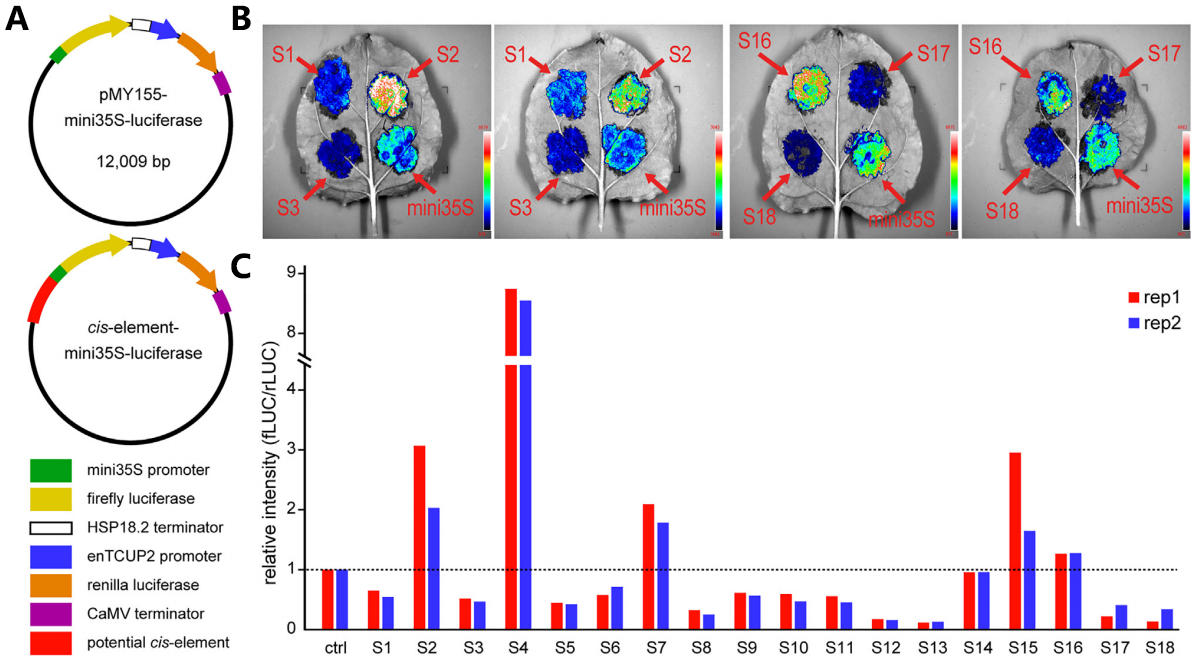
图9 验证类沉默子的转录抑制功能(Ouyang et al., 2023 )。(A) 验证沉默子的双荧光素酶报告系统;(B)冷CCD相机拍摄到的图片,根据荧光强度片段预测的沉默子是否具有转录抑制功能 ;(C) 基于萤火虫荧光素酶和海肾荧光素酶的比值(fLUC/rLUC)对18种预测沉默子的调节能力进行定量分析。
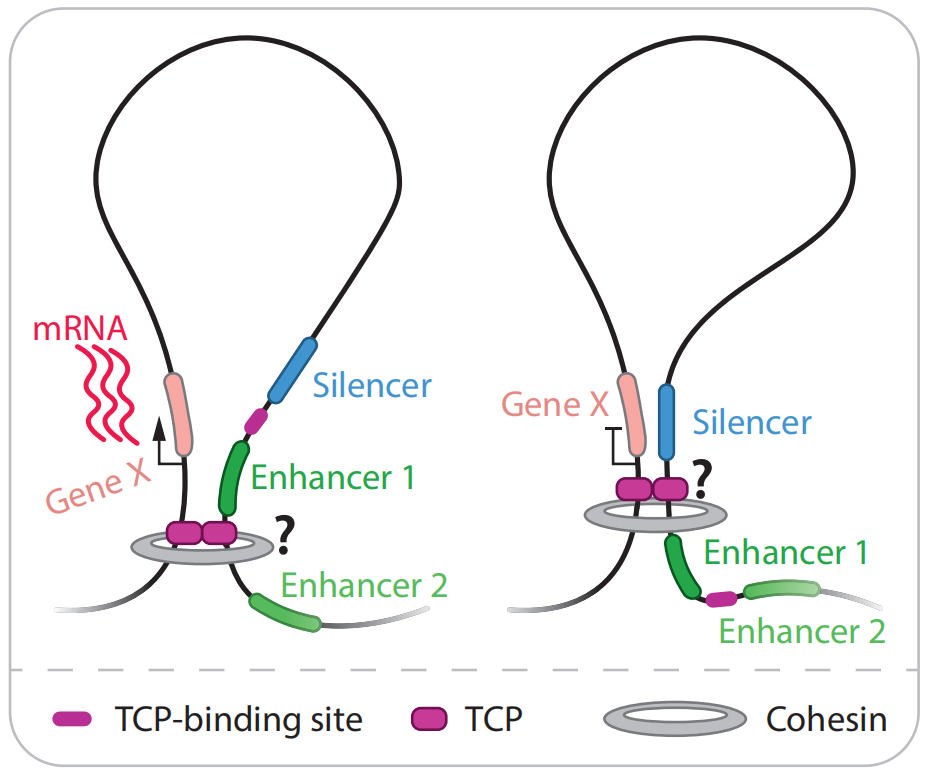
研究者利用长读型ChIA-PET绘制H3K27me3关联的三维基因组图谱来分析类沉默子关联的染色质相互作用情况。结果显示,类沉默子可以通过与远端的功能基因发生互作形成染色质环,从而抑制远端功能基因的表达。
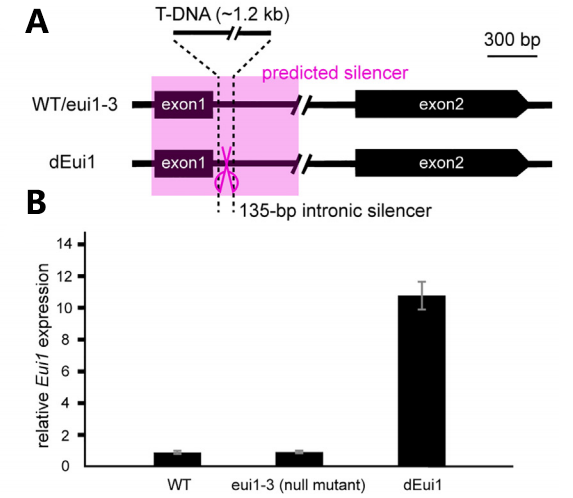
为了进一步验证类沉默子通过染色质相互作用能够抑制远端基因,研究者在预测的增强子中插入了T-DNA,创制了T-DNA插入突变体,对突变体(dEui1)、野生型、无效突变体(eui1-3)中远端基因的表达水平进行检测,结果显示类增强子发生缺失突变会引起与其互作的远端功能基因的转录上调(图10B),进一步验证了沉默子的转录抑制功能。
2、利用双荧光素酶报告基因实验(Dual-LUC)可以验证沉默子的转录抑制功能;
3、可通过ChIA-PET绘制H3K27me3关联的三维基因组图谱来分析类沉默子关联的染色质相互作用情况;
4、构建缺失突变体,检测突变体相比于野生型,受沉默子调控的基因的转录水平是否降低,来验证沉默子的功能。

Adiconis X, Haber A L, Simmons S K, et al. Comprehensive comparative analysis of 5′-end RNA-sequencing methods[J]. Nature methods, 2018, 15(7): 505-511.
Cai Y, Zhang Y, Loh Y P, et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions[J]. Nature communications, 2021, 12(1): 719.
Concia L, Veluchamy A, Ramirez-Prado J S, et al. Wheat chromatin architecture is organized in genome territories and transcription factories[J]. Genome biology, 2020, 21: 1-20.
Dong Q, Li N, Li X, et al. Genome‐wide Hi‐C analysis reveals extensive hierarchical chromatin interactions in rice[J]. The Plant Journal, 2018, 94(6): 1141-1156.
Gómez-Zambrano Á, Merini W, Calonje M. The repressive role of Arabidopsis H2A. Z in transcriptional regulation depends on AtBMI1 activity[J]. Nature Communications, 2019, 10(1): 2828.
Haberle V, Stark A. Eukaryotic core promoters and the functional basis of transcription initiation[J]. Nature reviews Molecular cell biology, 2018, 19(10): 621-637.
Inada DC, Bashir A, Lee C, et al. Conserved noncoding sequences in the grasses[J]. Genome Res, 2003, 13:2030–41.
Marand A P, Eveland A L, Kaufmann K, et al. cis-Regulatory elements in plant development, adaptation, and evolution[J]. Annual review of plant biology, 2023, 74: 111-137.
Meng F, Zhao H, Zhu B, et al. Genomic editing of intronic enhancers unveils their role in fine-tuning tissue-specific gene expression in Arabidopsis thaliana[J]. The Plant Cell, 2021, 33(6): 1997-2014.
Oka R, Zicola J, Weber B, et al. Genome-wide mapping of transcriptional enhancer candidates using DNA and chromatin features in maize[J]. Genome biology, 2017, 18: 1-24.
Ouyang W, Zhang X, Guo M, et al. Haplotype mapping of H3K27me3-associated chromatin interactions defines topological regulation of gene silencing in rice[J]. Cell Reports, 2023, 42(4).
Ricci W A, Lu Z, Ji L, et al. Widespread long-range cis-regulatory elements in the maize genome[J]. Nature plants, 2019, 5(12): 1237-1249.
Sieburth LE, Meyerowitz EM. Molecular dissection of the AGAMOUS control region shows that cis elements for spatial regulation are located intragenically[J]. Plant Cell, 1997, 9:355–65.
Xiao J, Jin R, Yu X, et al. Cis and trans determinants of epigenetic silencing by Polycomb repressive complex 2 in Arabidopsis[J]. Nature genetics, 2017, 49(10): 1546-1552.
Yan W, Chen D, Schumacher J, et al. Dynamic control of enhancer activity drives stage-specific gene expression during flower morphogenesis[J]. Nature communications, 2019, 10(1): 1705.
