植物的基因组中包含大量具有重要作用的功能元件,主要与农艺性状和作物驯化有关,靶向编辑这些功能元件可以精准改变重要农艺性状相关基因的表达,但目前植物表观基因组的资源还比较匮乏,科学家们认为向国际植物科学界发起植物ENCODE项目(pENCODE)是十分有必要的。那什么是ENCODE?以及建立pENCODE有什么意义呢?今天伯小远就给大家介绍一下ENCODE、ENCODE使用的技术方法以及我国科学家建立的小型pENCODE数据库。
3)从2013年至2016年,Phase 3扩大研究范围并增加了新型检测方法(图2),诸如通过配对末端标记(ChIA-PET)和Hi-C染色体构象捕获的染色质相互作用分析等方法揭示了染色质3D结构的特征。
4)从2017年至2021年,Phase 4,也称ENCODE 4,通过研究更广泛的生物样本(包括与疾病相关的生物样本),以及通过采用以前未在ENCODE中使用的新测定法,扩大人类和小鼠基因组中候选调控元件的目录。为了研究ENCODE已经编译的候选调控元件的生物学功能,ENCODE 4中添加了一个新的组成部分,即功能元件表征。
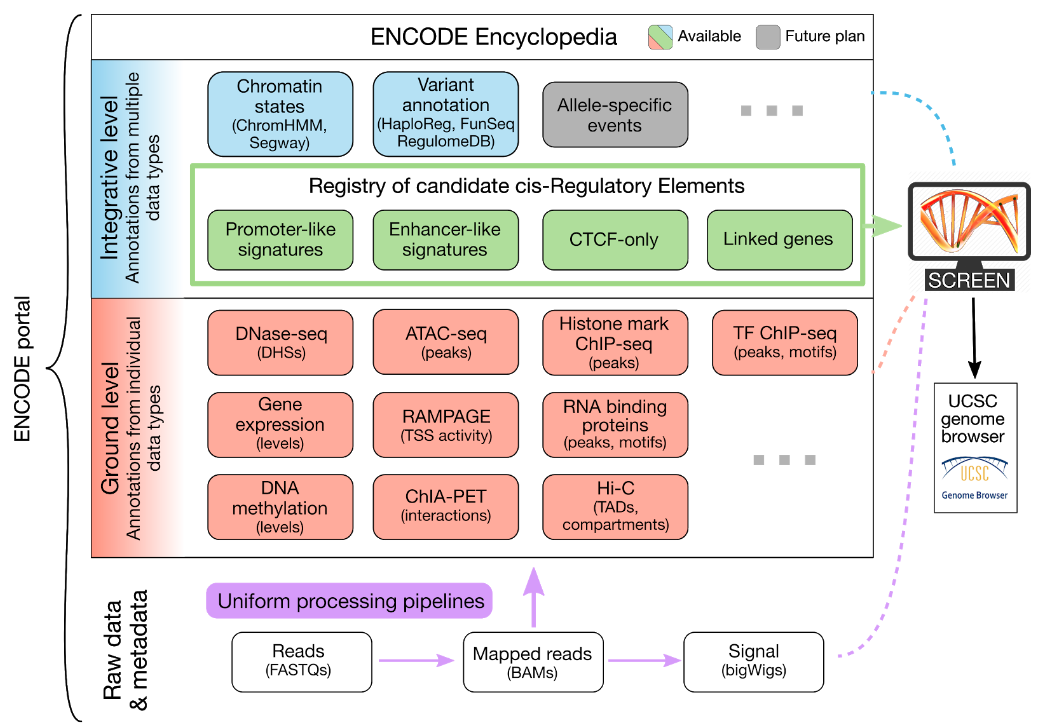
ENCODE项目联合会为了最大限度地提高研究者对ENCODE数据的访问权限,设立了超大型数据调度中心(Data Coordination Center,DCC),DCC的任务主要是和数据分析中心(Data Analysis Center)DAC合作定义数据类型,并将数据存储在数据库里,分享给全世界的研究人员。并且该项目能够协调全球各个实验室正在进行的研究,把研究重点放在一系列高度优先项目上,并使样本或数据的收集、获取和传播标准化。
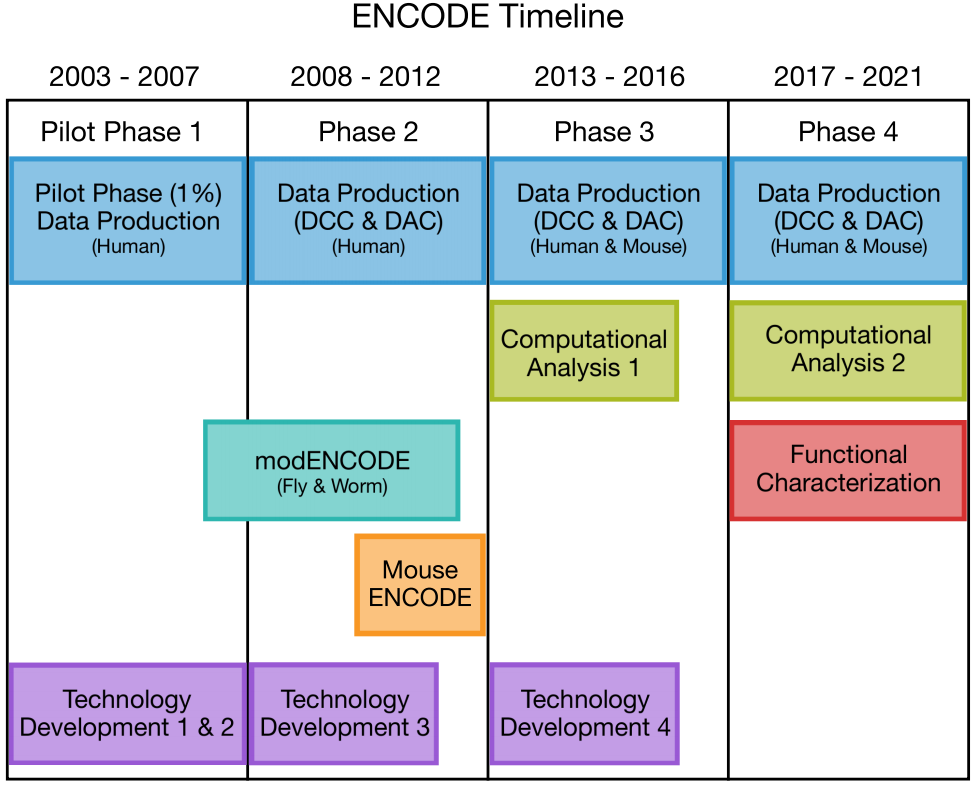
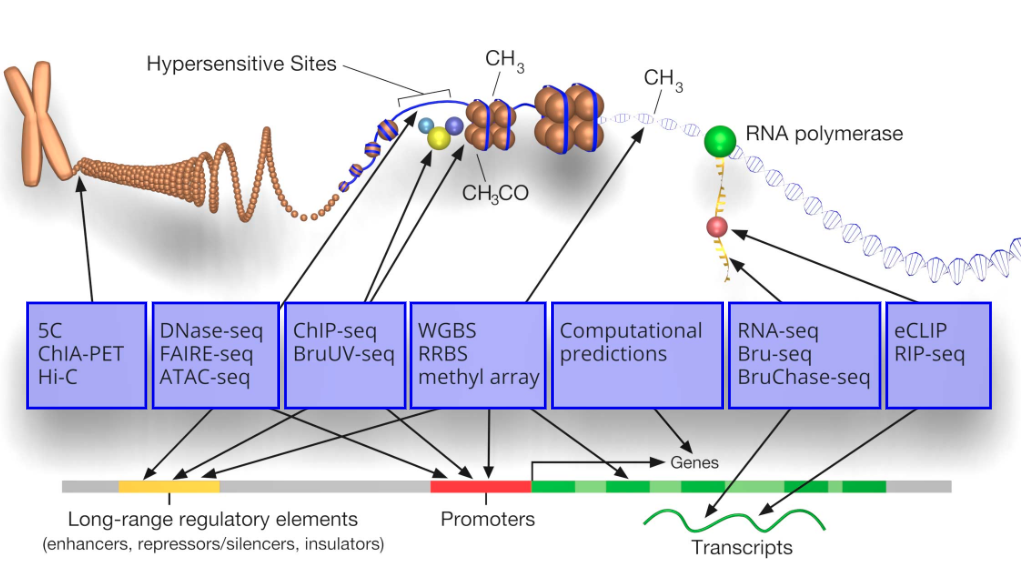
 图4 研究3D染色质结构技术(3C、4C、5C、Hi-C、ChIP-loop、ChIA-PET)的原理图(Hakim and Misteli, 2012)。
图4 研究3D染色质结构技术(3C、4C、5C、Hi-C、ChIP-loop、ChIA-PET)的原理图(Hakim and Misteli, 2012)。
这次主要介绍5C、Hi-C和ChIA-PET,这些技术源于3C(Chromosome conformation capture),3C的原理(图4):
(1)甲醛交联:分离完整的细胞核并进行甲醛交联,使参与染色质相互作用的蛋白固定;
(2)酶切序列:用限制性内切酶切割交联后的基因组;
(3)连接:连接酶连接互作片段,形成环状;
(4)解交联:将连接DNA片段的蛋白质消化掉,得到连接的DNA片段;
(5)PCR检测:使用位点特异性引物通过聚合酶链式反应(PCR)检测和定量单个连接产物。
Hi-C(High-throughput/resolution chromosome conformation capture)在3C的技术上进行了改良,限制酶切割后得到的片段具有平末端或粘性末端,然后将末端补平修复。利用末端修复机制,引入生物素标记的碱基。再进行连接处理,距离较近的DNA末端联结在一起。解交联后利用链亲和素磁珠捕获生物素,可富集含有互作关系的DNA片段,再进行文库构建,使用双末端法进行测序(图5)。Hi-C可以实现全基因组覆盖检测全部未知互作区域(Lieberman-Aiden et al., 2009)。
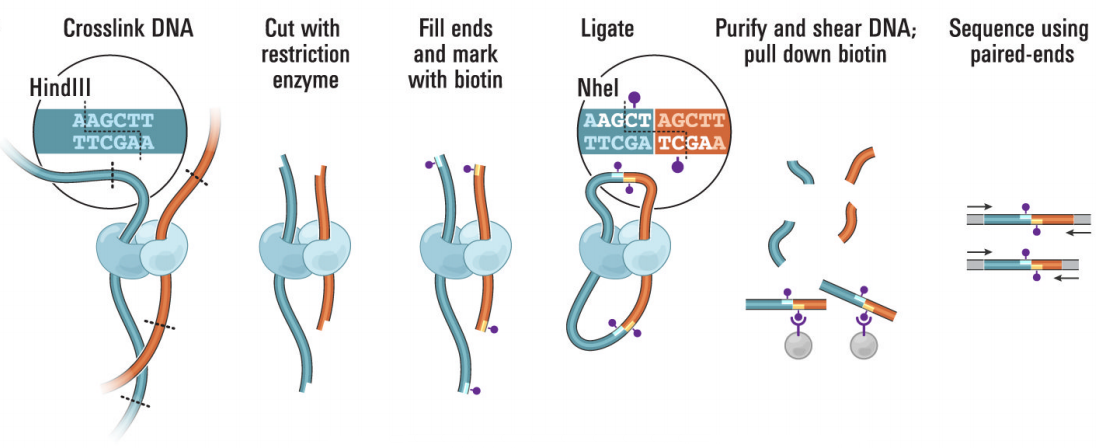
图5 Hi-C技术流程图(Lieberman-Aiden et al., 2009)。
ChIA-PET(Chromatin Interaction Analysis using Paired End Tag sequencing)将Hi-C与ChIP-seq结合,即在片段化DNA后使用特异性蛋白抗体富集DNA和蛋白质复合物,可以检测目的蛋白质的所有相互作用,同时该技术为了区分来自不同蛋白交联区域的序列,引入了双末端标签(Paired-End Tags)标记(图6)。双末端标签的目的在于区分连接之后的DNA序列是否来自同一个蛋白结合的序列(Li et al., 2019)。
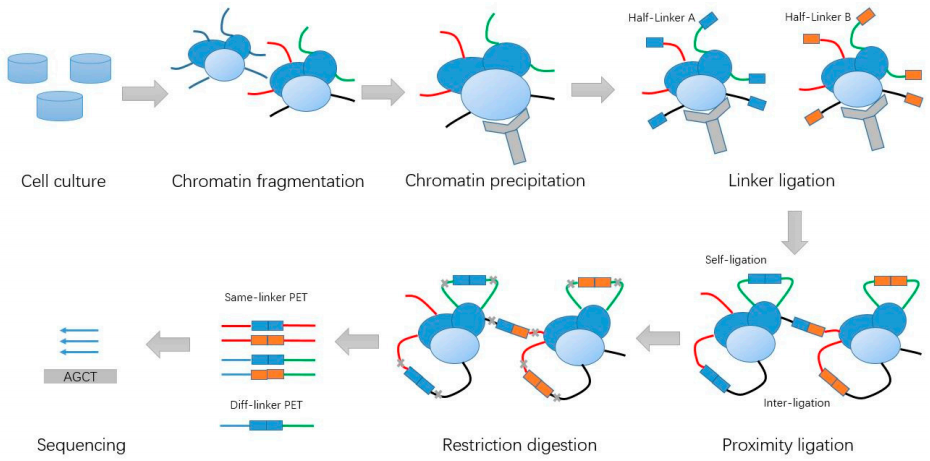
图6 ChIA PET实验步骤示意图(Li et al., 2019)。
这些技术在原理、覆盖度、测序方法以及局限性等方面的区别:
表1 3C、4C、5C、Hi-C、ChIP-loop、ChIA-PET的区别(Hakim and Misteli 2012)。
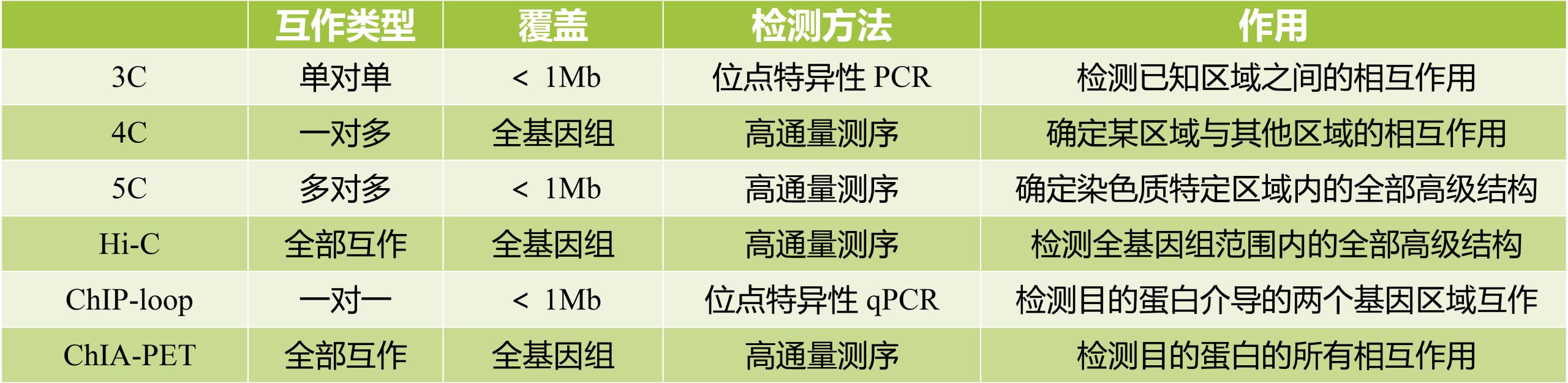


图9 已发表的植物基因组(截止到2020年12月)(Sun et al., 2022)。(A)自2000年拟南芥基因组发表以来,在染色体和非染色体水平上测序的植物基因组数量;(B)被子植物基因组测序最多的前10个家族;(C)被子植物多倍体基因组的测序数量。
植物基因组的调控区具有大量的农艺性状和作物驯化关联元件,靶向编辑这些调控元件可以精准改变重要农艺性状相关基因的表达。因此,建立植物表观组参考图谱将为作物遗传精准改良提供重要的资源。然而目前植物表观基因组的资源还比较匮乏。随着测序通量的不断提高,以及获取数据的便利,向国际植物科学界发起植物ENCODE项目(pENCODE)是十分有必要的(Lane et al., 2014)。

 2018年香港中文大学钟思林课题组在Nature Plants发表了题为“Genome encode analyses reveal the basis of convergent evolution of fleshy fruit ripening”的研究论文(Lu et al., 2018)。利用乙烯调节肉质果实的成熟在被子植物中非常普遍,这说明这些植物存在很明显的趋同进化过程,但其分子基础在很大程度上仍然未知。钟思林课题组开发了fruitENCODE项目,旨在采用ENCODE计划的方法,对七种跃变型(也称呼吸跃变型)肉质水果(苹果、香蕉、甜瓜、木瓜、桃、梨和番茄)的功能元件进行全面注释。还包括四种非跃变型肉质水果(黄瓜、葡萄、草莓和西瓜)和两种干果植物(拟南芥和水稻)进行比较分析。为了构建多肉果实功能基因组学的多维数据集,研究者使用BS-seq、ChIP-Seq、DNase-Seq和RNA-Seq等技术分别对其组织特异性DNA甲基化、组蛋白修饰、染色质可及性和转录组图谱进行了分析。还对由361个转录组、71个可及性染色质、147个组蛋白和45个DNA甲基化图谱组成的fruitENCODE数据进行分析,揭示了控制乙烯依赖性果实成熟的三种类型的转录调控通路,即MADS-type,NAC-type和dual-loop type(图10)。这些调控通路是由祖先被子植物的衰老或花器官身份途径通过新功能化或重新利用先前存在的基因进化而来的。
2018年香港中文大学钟思林课题组在Nature Plants发表了题为“Genome encode analyses reveal the basis of convergent evolution of fleshy fruit ripening”的研究论文(Lu et al., 2018)。利用乙烯调节肉质果实的成熟在被子植物中非常普遍,这说明这些植物存在很明显的趋同进化过程,但其分子基础在很大程度上仍然未知。钟思林课题组开发了fruitENCODE项目,旨在采用ENCODE计划的方法,对七种跃变型(也称呼吸跃变型)肉质水果(苹果、香蕉、甜瓜、木瓜、桃、梨和番茄)的功能元件进行全面注释。还包括四种非跃变型肉质水果(黄瓜、葡萄、草莓和西瓜)和两种干果植物(拟南芥和水稻)进行比较分析。为了构建多肉果实功能基因组学的多维数据集,研究者使用BS-seq、ChIP-Seq、DNase-Seq和RNA-Seq等技术分别对其组织特异性DNA甲基化、组蛋白修饰、染色质可及性和转录组图谱进行了分析。还对由361个转录组、71个可及性染色质、147个组蛋白和45个DNA甲基化图谱组成的fruitENCODE数据进行分析,揭示了控制乙烯依赖性果实成熟的三种类型的转录调控通路,即MADS-type,NAC-type和dual-loop type(图10)。这些调控通路是由祖先被子植物的衰老或花器官身份途径通过新功能化或重新利用先前存在的基因进化而来的。
fruitENCODE的测序数据存储在NCBI SRA数据库,登录号为PRJNA381300,经处理的数据存储在NCBI GEO数据库,登录号为GSE116581。
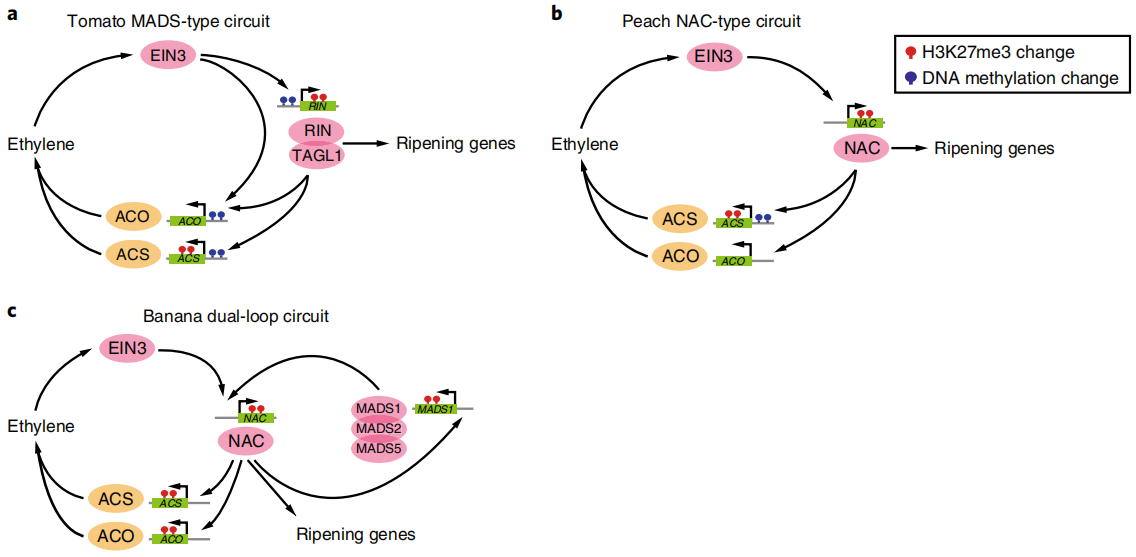
图10 控制跃变型果实成熟的三种转录调控通路(Lu et al., 2018)。(a)番茄果实成熟调控模型。乙烯转录因子EIN3激活MADS转录因子RIN。RIN与TOMATO AGAMOUS-LIKE1(TAGL1)形成复合物,并激活乙烯生物合成基因,形成正反馈调控通路,在成熟过程中产生自催化乙烯。下游成熟基因通过MADS转录因子直接偶联到这条调控通路上。在叶片和未成熟果实中,该调控通路会受到与启动子DNA超甲基化和抑制性组蛋白H3K27me3相关的关键基因抑制;(b)桃果实成熟调节模型,利用NAC而不是MADS转录因子;(c)香蕉果实成熟调节模型。NAC和MADS之间的额外通路使香蕉果实能够在乙烯抑制剂1-甲基环丙烯(MCP)的存在下合成乙烯。

水稻(Oryza sativa L.)是我国乃至全世界重要的粮食作物,同时也是基础研究的重要模式植物。水稻基因组DNA顺式调控元件的注释和鉴定,对理解水稻基因表达调控的机理有重要意义。2021年华中农业大学李国亮教授和李兴旺教授带领的团队在Molecular Plant发表了题为“RiceENCODE:A comprehensive epigenomic database as a rice Encyclopedia of DNA Elements”的研究论文(Xie et al., 2021)。研究者们建立了一个水稻多元表观基因组数据的数据库RiceENCODE(http:/glab.hzau.edu.cn/RiceENCODE/),使用包括ChIP-seq、FAIRE-seq、MNase-seq、ATAC-seq、ncRNA-seq、RNA-seq,Hi-C和ChIA-PET等技术检测,共计972套水稻高通量组学数据,涉及三维染色质相互作用、组蛋白修饰、染色质状态、染色质可及性、DNA甲基化和转录组信息。通过标准化的数据处理流程,得到了多维度的高质量表观和三维基因组数据(图11)。这极大地方便了研究人员查询和分析水稻的表观遗传信息,促进了水稻表观和三维基因组的研究。
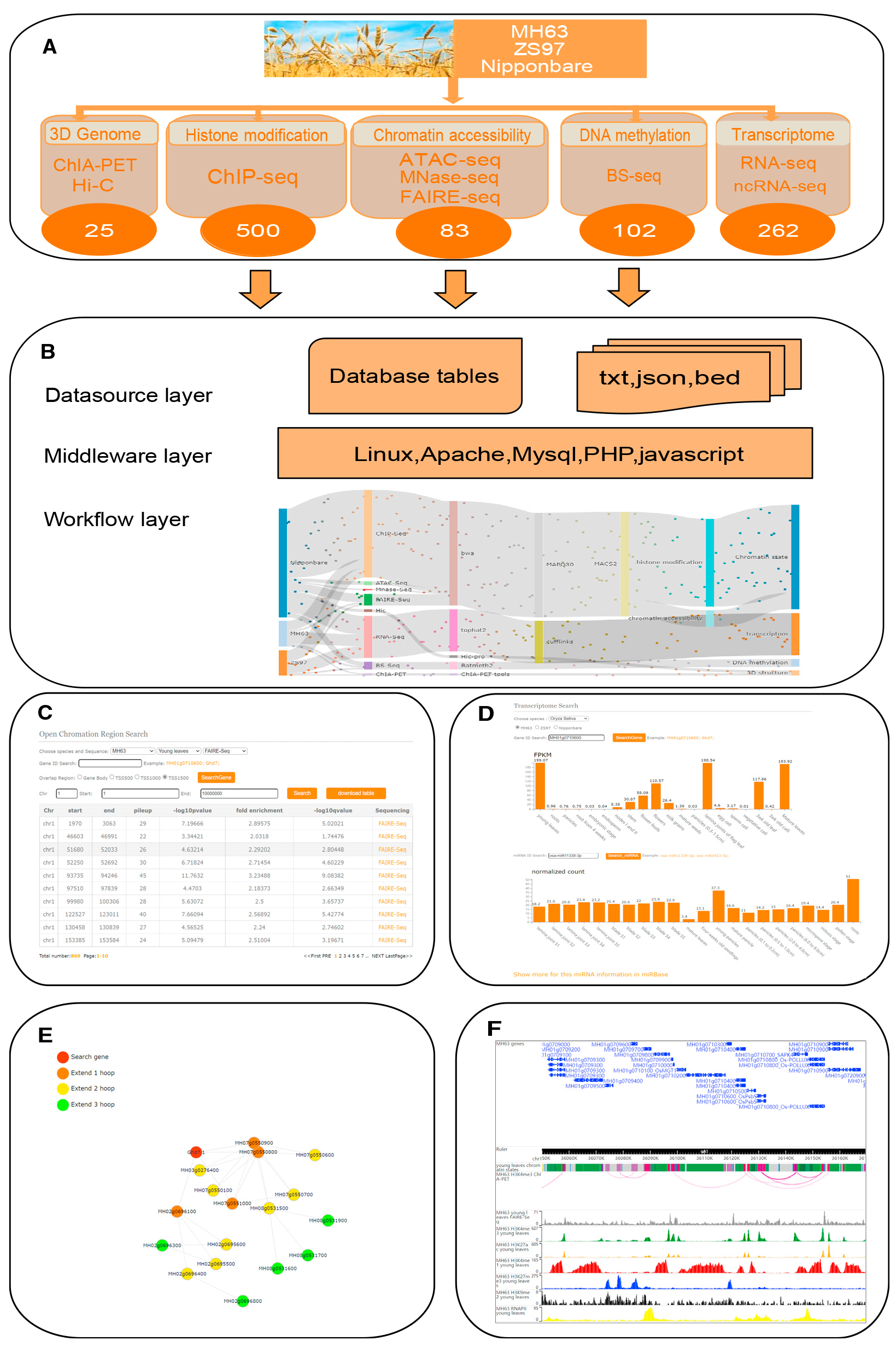
图11 RiceENCODE数据库的体系结构(Xie et al., 2021)。(A)RiceENCODE数据库的组成;(B)RiceENCODE的网站架构;(C)染色质可及性数据库;(D)mRNA和miRNA表达水平数据库;(E)基因互作数据库;(F)WashU表观基因组可视化展示。

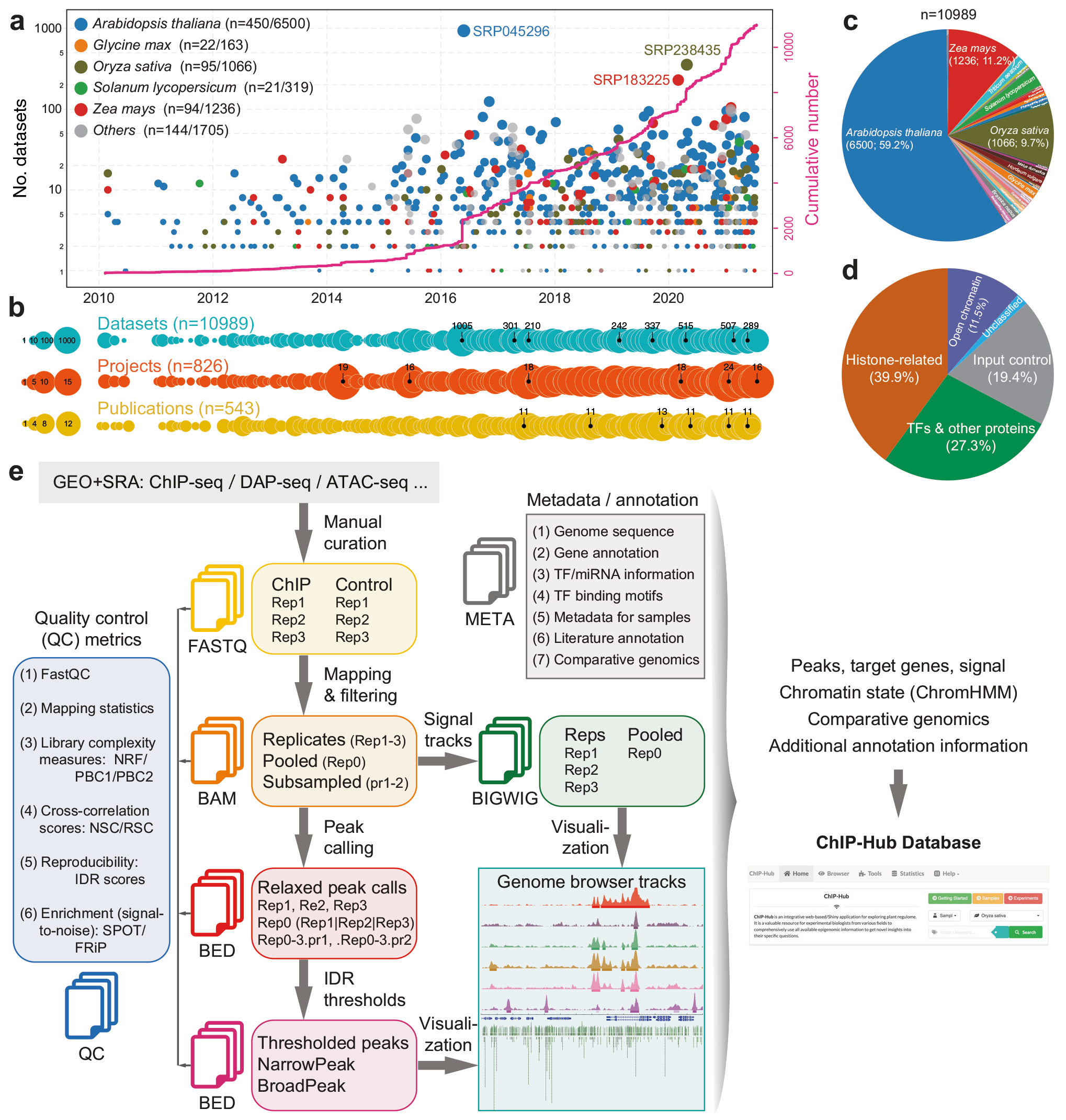
2022年,南京大学陈迪俊课题组在Nature Communications杂志上发表了题为“ChIP-Hub provides an integrative platform for exploring plant regulome”的文章(Fu et al., 2022)。他们从NCBI SRA数据库中收集来自世界各地不同实验的超过40多个植物物种,总量大于10000个的公开的调控组数据集。这些数据都是通过高通量测序实验产生的,包括ChIP-seq、DAP-seq、DNase-seq和ATAC-seq。陈迪俊课题组基于ENCODE标准以统一的方式对其进行重新分析,并将数据资源和分析结果整合到ChIP-Hub(https://biobigdata.nju.edu.cn/ChIPHub/)在线数据库中,可用于可视化和多组学分析(图12)。
Consortium EP, Birney E, Stamatoyannopoulos JA, et al. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799-816.
Consortium EP, Snyder MP, Gingeras TR, et al. 2020. Perspectives on ENCODE. Nature 583:693-698.
Dekker J, Rippe K, Dekker M, et al. 2002. Capturing chromosome conformation. Science 295:1306-1311.
Dostie J, Richmond TA, Arnaout RA, et al. 2006. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 16:1299-1309.
Fu LY, Zhu T, Zhou X, et al. 2022. ChIP-Hub provides an integrative platform for exploring plant regulome. Nat Commun 13:3413.
Hakim O, Misteli T. 2012. SnapShot: Chromosome confirmation capture. Cell 148:1068 e1061-1062.
Lane AK, Niederhuth CE, Ji L, et al. 2014. pENCODE: a plant encyclopedia of DNA elements. Annu Rev Genet 48:49-70.
Li G, Sun T, Chang H, et al. 2019. Chromatin Interaction Analysis with Updated ChIA-PET Tool (V3). Genes (Basel) 10.
Lieberman-Aiden E, van Berkum NL, Williams L, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289-293.
Lu P, Yu S, Zhu N, et al. 2018. Genome encode analyses reveal the basis of convergent evolution of fleshy fruit ripening. Nat Plants 4:784-791.
Stockwell PA, Chatterjee A, Rodger EJ, et al. 2014. DMAP: differential methylation analysis package for RRBS and WGBS data. Bioinformatics 30:1814-1822.
Sun Y, Miao N, Sun T. 2019. Detect accessible chromatin using ATAC-sequencing, from principle to applications. Hereditas 156:29.
Sun Y, Shang L, Zhu QH, et al. 2022. Twenty years of plant genome sequencing: achievements and challenges. Trends Plant Sci 27:391-401.
Xie L, Liu M, Zhao L, et al. 2021. RiceENCODE: A comprehensive epigenomic database as a rice Encyclopedia of DNA Elements. Mol Plant 14:1604-1606.
