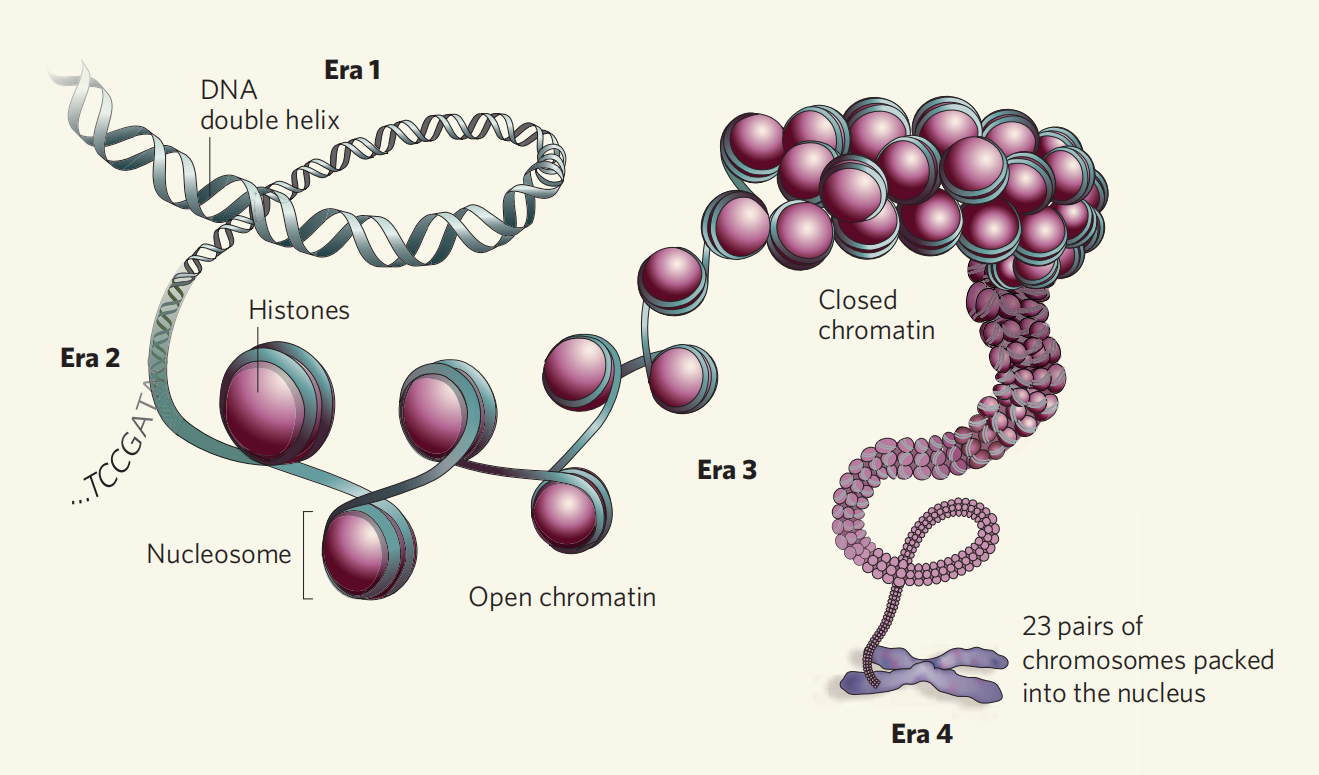
一般来说,调控元件选择性地定位于可及性染色质,这对转录调控至关重要(Thurman et al., 2012)。虽然转录因子的占用并不一定与染色质可及性呈正相关(Hsiung et al., 2015),但维持染色质可及性的构型需要与转录因子结合来激活靶基因(Morris et al., 2014)(图1)。另一方面,异染色质(或被称为封闭染色质),限制了转录因子和转录调控因子与启动子或增强子的结合,从而导致基因沉默(Stergachis et al., 2013;Rinn J L and Chang H Y., 2012;Chen T and Dent S Y R, 2014)(图1)。简单地讲,就是基因的转录需要将DNA的高级结构解开,但不需要DNA链全部解开,只需要打开一部分,也就是将表达基因的区域解开即可,而这一过程,主要由染色体组蛋白修饰(尤其是乙酰化)来实现。这部分打开的染色质,就叫开放染色质。而染色质一旦打开,就允许一些调控蛋白(比如转录因子)跑过来与之相结合。
染色质的可及性是表观遗传学的重要组成部分,除此之外,表观遗传学还包括核小体定位、转录因子占用等,对表观遗传学的研究主要采用高通量、全基因组的方法,例如:MNase-seq、DNase-seq、FAIRE-seq、ATAC-seq等。
1、目前的方法会平均和“掩盖”细胞群的异质性;
2、细胞通常必须在体外生长才能获得足够的物质,而这样的条件会扰乱体内环境,并以未知的方式调节表观遗传状态。
科学家们为了克服当前方法的局限性,就开发出了新的技术对表观遗传的整体面貌进行分析。ATAC-Seq就是在这种情况下诞生的!
ATAC-seq是“Assay for Transposase-Accessible Chromatin with high-throughput Sequencing”的缩写,最早是由斯坦福大学的Howard Chang和William Greenleaf实验室的首席研究员Jason Buenrostro于2013年在Nature Methods首次发表。
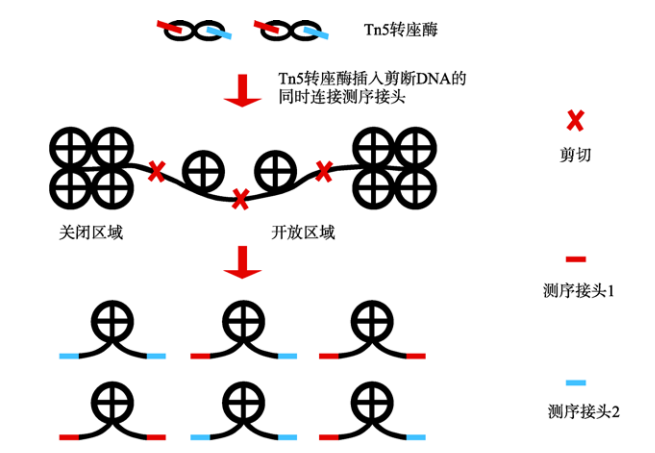
通俗地讲,就是Tn5转座酶可以随机结合并切割染色质开放区的DNA,并且可同时在切割位点插入接头序列。只要将携带已知DNA序列标签的转座复合物(即带红色和绿色序列标签的Tn5转座酶)加入到细胞核中一起孵育,再利用已知序列标签进行PCR扩增即可形成文库,经过测序就可以获得染色质开放区的信息了。
1、目前的方法会平均和“掩盖”细胞群的异质性;
2、细胞通常必须在体外生长才能获得足够的物质,而这样的条件会扰乱体内环境,并以未知的方式调节表观遗传状态。
科学家们为了克服当前方法的局限性,就开发出了新的技术对表观遗传的整体面貌进行分析。ATAC-Seq就是在这种情况下诞生的!
ATAC-seq是“Assay for Transposase-Accessible Chromatin with high-throughput Sequencing”的缩写,最早是由斯坦福大学的Howard Chang和William Greenleaf实验室的首席研究员Jason Buenrostro于2013年在Nature Methods首次发表。
通俗地讲,就是Tn5转座酶可以随机结合并切割染色质开放区的DNA,并且可同时在切割位点插入接头序列。只要将携带已知DNA序列标签的转座复合物(即带红色和绿色序列标签的Tn5转座酶)加入到细胞核中一起孵育,再利用已知序列标签进行PCR扩增即可形成文库,经过测序就可以获得染色质开放区的信息了。
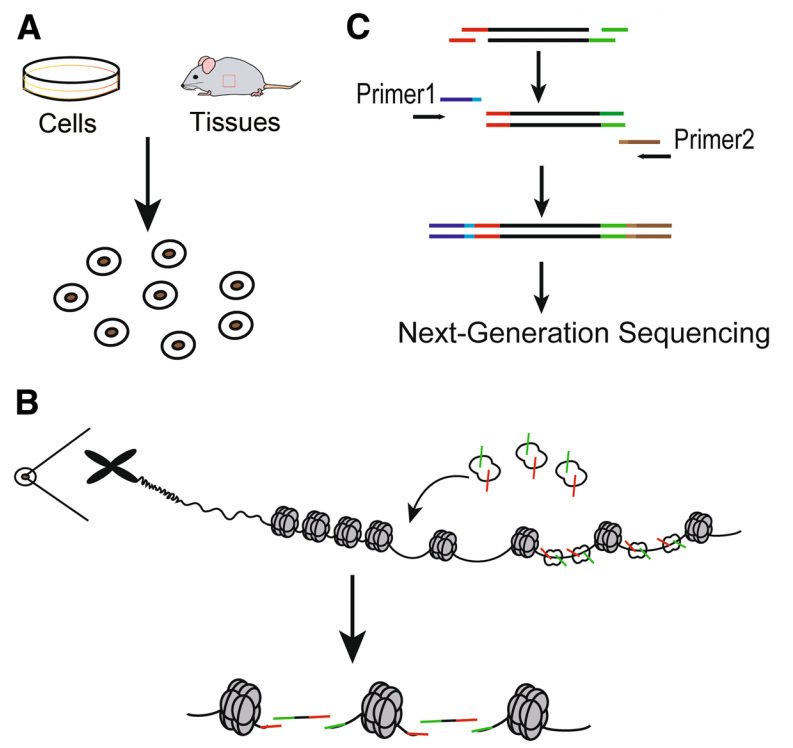
注:这里的实验过程虽然是以动物实验为例进行说明的,但是同样适用于植物。
在测序前需要对ATAC-seq文库进行质量控制,从而保证文库浓度达到测序标准。对符合条件的文库进行测序,收集原始reads。通过对测序数据进行质量评估,以及对数据进行过滤后,进一步获得干净的reads(Davie et al., 2015;Bell et al., 2011;Mardis E R, 2008)。去除标签引物序列和低质量reads后,处理长度约150个核苷酸(nts)的高质量reads,进行进一步分析(Miskimen et al., 2017)。Peak calling reads被映射到参考基因组和可及性的染色质区域,如启动子、增强子和绝缘子(Kumasaka et al., 2016;Ackermann et al., 2016;Quillien et al., 2017)。可以进一步进行一系列详细地分析,如确定全基因组reads的分布,确定峰长的分布,对具有识别峰的基因进行功能分析,基因功能元件上的峰的分布,以及样本间差异峰的分析等(Pranzatelli et al., 2018;Mu et al., 2012)。
ATAC-seq技术的优缺点
表1 几种测序方法比较(Sun et al., 2019)。

对于得到的结果,从下图中可以看出ATAC-seq出来的结果,和传统方法出来的结果具有很强的一致性。
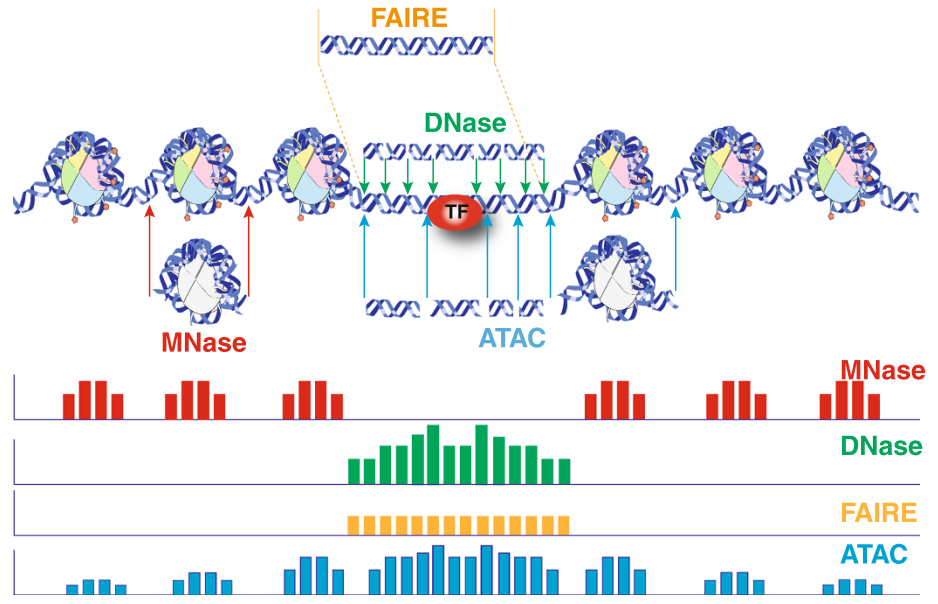
注:从图中可以看出,TF与裸露染色质的结合会降低染色质区域的开放性。DNase-seq和ATAC-seq技术都能够识别这一信息,在它的峰中会出现一个轻微的凹槽——也就是足迹(Footprint)。但由于ATAC-seq技术的限制(当转录因子结合DNA时,会阻止Tn5酶转座酶在该点上的切割,所以会形成一个保护区域,reads无法富集到中间的部分),凹槽的辨识度不高,难以用来做足迹分析,也就是TF与染色质的结合研究。
2、简化的实验过程减少了样品制备的时间,减少了误差的概率,显著提高了实验的成功率和重复性。相比而言,DNase-seq和MNase-seq实验通常需要2-3天,FAIRE-seq实验通常需要3-4天。
3、样本量至少减少了1000倍,从100万个细胞(FAIRE-sesq)和5000万个细胞(DNase-seq)减少到大约500个细胞。当样本采集具有挑战性时,这一优势尤为突出。
4、ATAC-seq利用双端测序技术绘制核小体的定位和占用图谱。双端测序可以对DNA片段的两端进行测序,使基因组重复区域的reads比对更加准确。
5、ATAC-seq的重复性,比MNase-seq和DNase-seq的更强,操作起来也更加简便,而且只需要很少的细胞/组织量,同时测序信号更加好。
6、单细胞测序技术是近几年研究的热点,通过对单细胞的测序能够将表观遗传学研究个体化。常见的ChIP-seq、DNase-seq、MNase-seq等技术无法进行单细胞测序,而ATAC-seq经过实验验证是可以进行单细胞测序的。
2、研究表明,没有核小体和转录因子的“裸”DNA更容易被Tn5转座酶剪切。此外,Tn5转座酶倾向于在转录因子结合区域进行结合和切割,从而导致部分转录因子信息的丢失。所有这些缺点使得ATAC-seq难以检测转录因子的足迹,而这可用于识别转录因子的潜在结合基序。
3、由于线粒体DNA的存在,通过ATAC-seq获得的数据不可避免地包含一些线粒体reads。根据细胞类型,ATAC-seq数据可能包含20-80%的线粒体测序reads。
ATAC-seq与ChIP-seq的区别
要看这两个技术之间的区别,先来看看它们之间的联系,这两种技术都可以用来研究转录因子与启动子的结合,但是这两个技术之间存在一些区别:ChIP-Seq技术在实验之前就有一个明确感兴趣的转录因子,也就是研究者需要对某个表观遗传学机制所起的作用有一个初步的想法,然后根据目标转录因子设计抗体去做ChIP实验拉到与其结合的DNA,验证感兴趣的转录因子是否与DNA存在相互作用;而ATAC-Seq技术没有落脚到具体哪个转录因子,也就是不涉及某个具体的表观遗传机制,是在全基因组范围内检测染色质的开放程度,可以得到全基因组范围内的蛋白质可能结合的位点信息,用这个技术方法与其他方法结合是想去筛感兴趣的调控因子。许多研究人员已开始使用ATAC-Seq作为初步筛查方法来鉴定样品之间染色质可及性区域的变化,然后据此进行后续实验。
通过上面的介绍相信大家对ATAC-seq这个技术了解的已经比较清楚了,那么这个技术有哪些方面的应用呢?下面我们先简单看看,后面再给大家结合文献具体去看!
关于ATAC-seq的介绍我们就先将讲这么多,相信大家看完这篇文章对ATAC-seq都会有一个清晰地认识,后面伯小远会结合具体的文献给大家讲一讲ATAC-seq的应用,感兴趣的同学可以继续关注哦!
References:
陈敏, 张峥, 孟紫媛,等. ATAC-seq在复杂疾病研究中的应用进展[J]. 遗传, 2020, 42(4):7.
Ackermann A M, Wang Z, Schug J, et al. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes[J]. Molecular metabolism, 2016, 5(3): 233-244.
Bártová E, Kozubek S. Nuclear architecture in the light of gene expression and cell differentiation studies[J]. Biology of the Cell, 2006, 98(6): 323-336.
Baylin S B, Schuebel K E. The epigenomic era opens[J]. Nature, 2007, 448(7153): 548-549.
Bell O, Tiwari V K, Thomä N H, et al. Determinants and dynamics of genome accessibility[J]. Nature Reviews Genetics, 2011, 12(8): 554-564.
Buenrostro J D, Giresi P G, Zaba L C, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position[J]. Nature methods, 2013, 10(12): 1213-1218.
Chen T, Dent S Y R. Chromatin modifiers and remodellers: regulators of cellular differentiation[J]. Nature Reviews Genetics, 2014, 15(2): 93-106.
Chuong E B, Elde N C, Feschotte C. Regulatory activities of transposable elements: from conflicts to benefits[J]. Nature Reviews Genetics, 2017, 18(2): 71-86.
Davie K, Jacobs J, Atkins M, et al. Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-seq and FAIRE-seq open chromatin profiling[J]. PLoS genetics, 2015, 11(2): e1004994.
Gottesfeld J M, Carey M F. Introduction to the thematic Minireview series: chromatin and transcription[J]. Journal of Biological Chemistry, 2018, 293(36): 13775-13777.
Haniford D B, Ellis M J. Transposons Tn10 and Tn5[J]. Mobile DNA III, 2015: 631-645.
Hsiung C C S, Morrissey C S, Udugama M, et al. Genome accessibility is widely preserved and locally modulated during mitosis[J]. Genome research, 2015, 25(2): 213-225.
Jackson D A. Nuclear organization: uniting replication foci, chromatin domains and chromosome structure[J]. Bioessays, 1995, 17(7): 587-591.
Kumasaka N, Knights A J, Gaffney D J. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq[J]. Nature genetics, 2016, 48(2): 206-213.
Mardis E R. Next-generation DNA sequencing methods[J]. Annual review of genomics and human genetics, 2008, 9(1): 387-402.
Miskimen K L S, Chan E R, Haines J L. Assay for Transposase‐Accessible Chromatin Using Sequencing (ATAC‐seq) Data Analysis[J]. Current Protocols in Human Genetics, 2017, 92(1): 20.4. 1-20.4. 13.
Morris S A, Baek S, Sung M H, et al. Overlapping chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions[J]. Nature structural & molecular biology, 2014, 21(1): 73-81.
Mu J C, Jiang H, Kiani A, et al. Fast and accurate read alignment for resequencing[J]. Bioinformatics, 2012, 28(18): 2366-2373.
Pederson T. The spatial organization of the genome in mammalian cells[J]. Current opinion in genetics & development, 2004, 14(2): 203-209.
Picelli S, Björklund Å K, Reinius B, et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects[J]. Genome research, 2014, 24(12): 2033-2040.
Pranzatelli T J F, Michael D G, Chiorini J A. ATAC2GRN: optimized ATAC-seq and DNase1-seq pipelines for rapid and accurate genome regulatory network inference[J]. BMC genomics, 2018, 19(1): 1-13.
Quillien A, Abdalla M, Yu J, et al. Robust identification of developmentally active endothelial enhancers in zebrafish using FANS-assisted ATAC-Seq[J]. Cell reports, 2017, 20(3): 709-720.
Reznikoff W S. The TN5 transposon[J]. Annual review of microbiology, 1993, 47(1): 945-963.
Rinn J L, Chang H Y. Genome regulation by long noncoding RNAs[J]. Annual review of biochemistry, 2012, 81.
Stergachis A B, Neph S, Reynolds A, et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes[J]. Cell, 2013, 154(4): 888-903.
Sun Y, Miao N, Sun T. Detect accessible chromatin using ATAC-sequencing, from principle to applications[J]. Hereditas, 2019, 156(1): 1-9.
Thurman R E, Rynes E, Humbert R, et al. The accessible chromatin landscape of the human genome[J]. Nature, 2012, 489(7414): 75-82.
Tsompana M, Buck M J. Chromatin accessibility: a window into the genome[J]. Epigenetics & chromatin, 2014, 7(1): 1-16.
