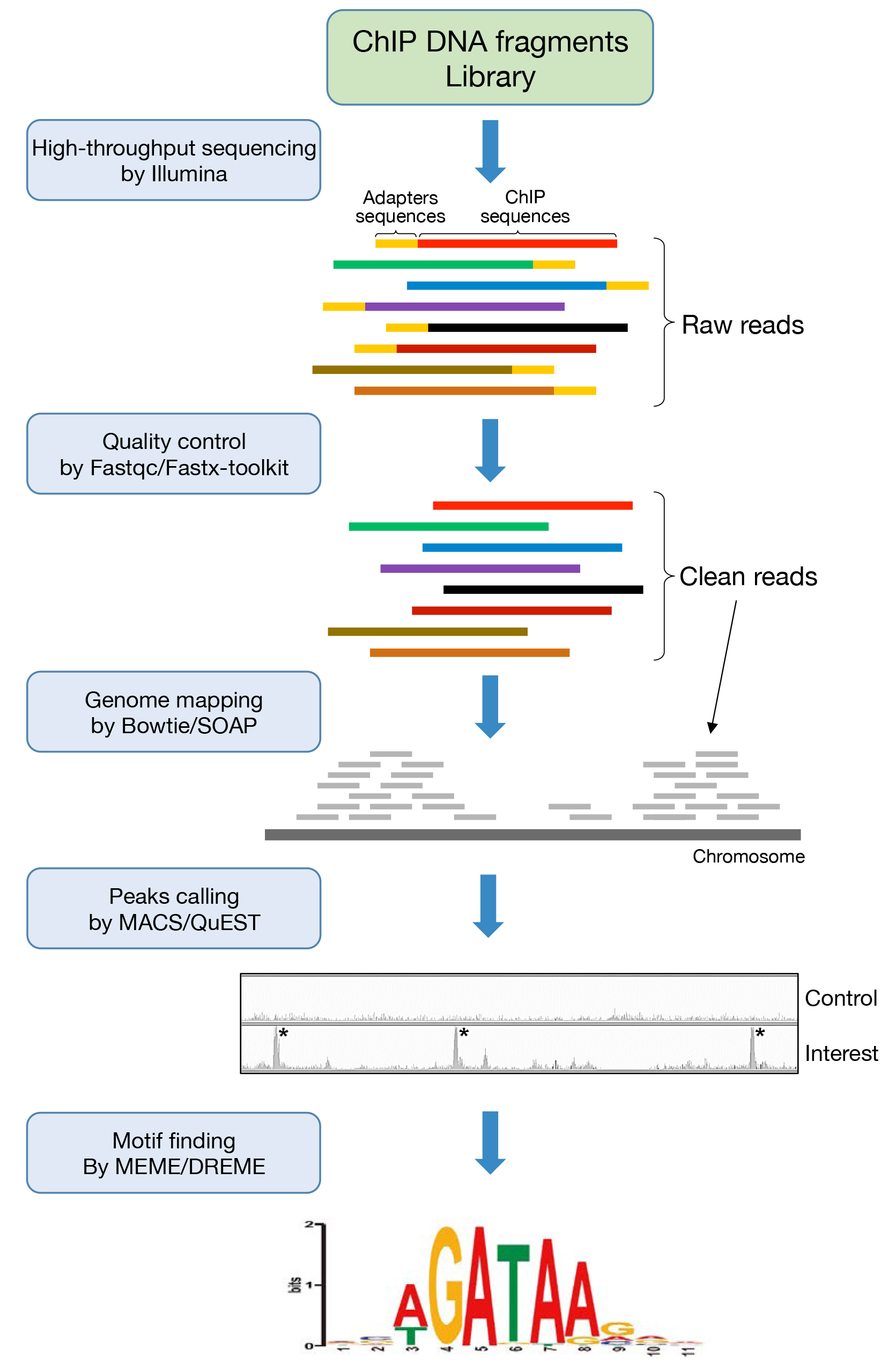
图1 ChIP-seq数据分析工作流程概述(Chen et al., 2018)。
这里给大家介绍几个专业名词:
Peak calling:查找DNA结合位点的步骤一般叫做peak calling。TF在基因组上的结合其实是一个随机过程,基因组的每个位置其实都有机会结合某个TF,只是概率不一样,说白了,peaks出现的位置,是TF结合的热点,而peak calling就是为了找到这些热点。如何定义热点呢?通俗地讲,热点是这样一些位置,这些位置多次被测得的reads所覆盖(测序时一般测的都是一个细胞群体,reads出现次数多,说明该位置被TF结合的几率大)。换句简单的话说,你可以理解peaks就是reads峰。
Peak注释:所谓的peaks注释,首先看peaks在基因组的哪一个区段,看看它们在基因不同区域(基因上下游,5’/3’-UTR,启动子,内含子)的分布情况。最典型的对于转录因子,通常都是位于基因的启动子区;其次是邻近的基因的注释,蛋白结合到DNA上之后,主要是发挥基因表达调控的功能,这些peaks区域附近的基因就作为其候选的调控基因。
Motif(基序)分析:在ChIP-seq数据分析中,motif分析是一项重要的分析内容。通过motif分析,我们可以对转录因子结合位点的序列模式有进一步的了解,那么什么是motif呢?蛋白质发挥功能的基本单元是domain,是一种特殊的三维结构,不同结构的domain与其他分子特异结合从而发挥功能。与此类似,转录因子在与DNA序列结合时,其结合位点的序列也由于一定的特异性,不同转录因子结合的DNA序列的模式是不同的。为了更好的描述结合位点序列的模式,科学家们提出了motif的概念——特定碱基序列的模式即为motif。
下面是我们在文章中或motif分析中经常见到的图,用来表征序列的一致性和多样性。字母越大,代表在该位置出现该核苷酸或者氨基酸的概率越大,常用Bits或者百分比表示。

为了加深大家对ChIP-seq的理解,下面再从实验本身的角度为大家讲解一下这个实验的过程,希望通过伯小远的反复讲解大家对这个实验可以有一个比较透彻的理解,等理解完这个实验,我们再去看它的应用!
DNA片段化:DNA片段化是获得良好的ChIP分辨率的关键因素,理想情况下的片段大小在200到1000bp之间。剪切是最难控制的步骤之一。通过超声处理和/或核酸酶/酶促消化可以实现剪切,各有利弊。超声处理虽然需要大量的手动操作,但非常适合难以裂解的细胞;酶促消化不需要手动操作,适用于大量样品的处理,但其剪切位点不是随机的。
免疫沉淀:抗体免疫沉淀,实验组加入预先结合了特异性抗体的beads进行孵育,最终形成beads-抗体-目的蛋白-DNA复合物。
解交联:洗脱得到抗体-目的蛋白-DNA复合物,去除非特异性结合的DNA片段,使用蛋白酶K/NaCl处理进行解交联,最后将DNA片段纯化回收。
验证:通过qPCR对ChIP结果进行验证。
高通量测序:准备好ChIP后的DNA样品用于ChIP-seq建库,质检及测序。
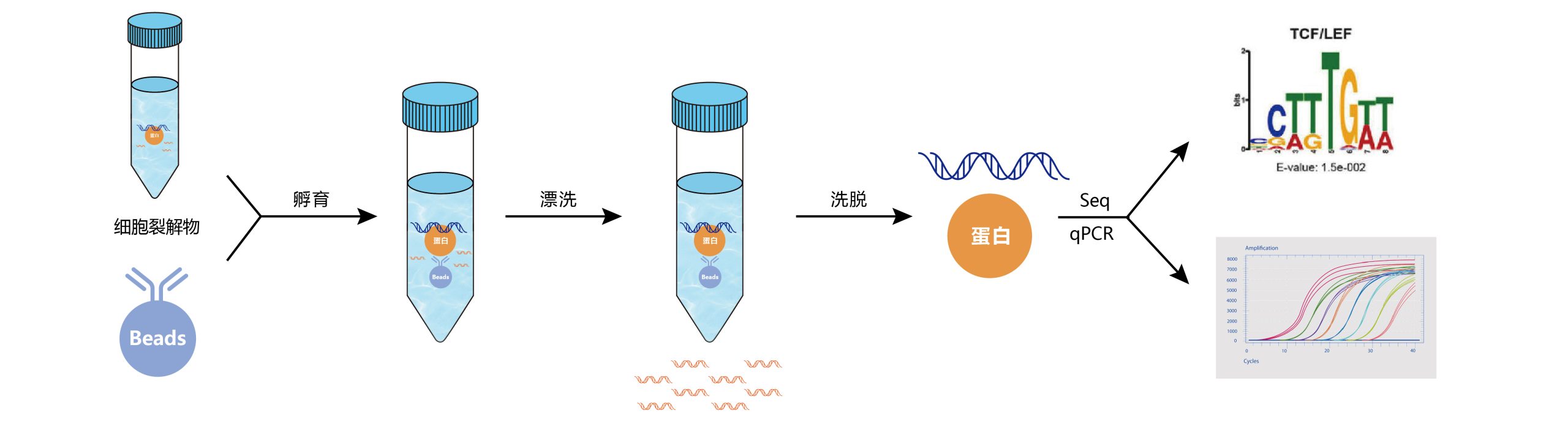
图3 染色质免疫沉淀ChIP-seq/qPCR实验流程。
2、 ChIP-seq可用来研究转录因子结合位点,解析该转录因子作用的通路信息;
3、ChIP-seq技术可得到核小体的定位图谱,核小体定位在转录调控,DNA复制和修复等多种细胞过程中并起着重要作用;
4、ChIP-seq技术可研究DNA的甲基化情况,DNA甲基化会引起染色体结构、DNA构象、DNA稳定性以及DNA与蛋白质相互作用方式的改变,从而控制基因表达。
针对ChIP-seq这4个方面的应用,为了让大家能更好地理解文献举例里面的内容,小远决定在这里先给大家把基础打牢,如果不清楚基本概念,那么有些实验结果可能会比较难理解,所以放慢脚步看看下面都有些什么内容需要我们提前学习吧!
核小体的结构如下:两个组蛋白H2A、H2B、H3和H4聚在一起形成一个组蛋白八聚体,它结合和包裹大约1.7圈的DNA,或约146个碱基对。一个H1蛋白的加入包裹了另外20个碱基对,导致八聚体发生了两次完整的旋转,形成了一个称为染色体的结构(图6中的Box 4)。考虑到平均每条染色体包含1亿多对DNA碱基对,由此产生的166个碱基对并不算长。因此,每条染色体包含数十万个核小体,这些核小体由它们之间的DNA连接(平均约20个碱基对)。这个连接的DNA被称为连接DNA。因此,每条染色体都是一个长链的核小体,当用电子显微镜观察时,它看起来像一串珠子(图4)(Olins & Olins, 1974, 2003)。
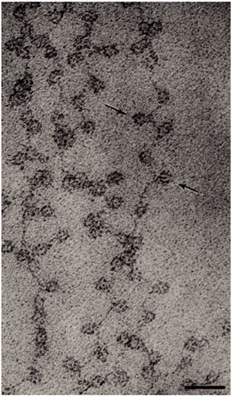
图5 染色质的电子显微图:串珠(Olins D E and Olins A L, 2003)。
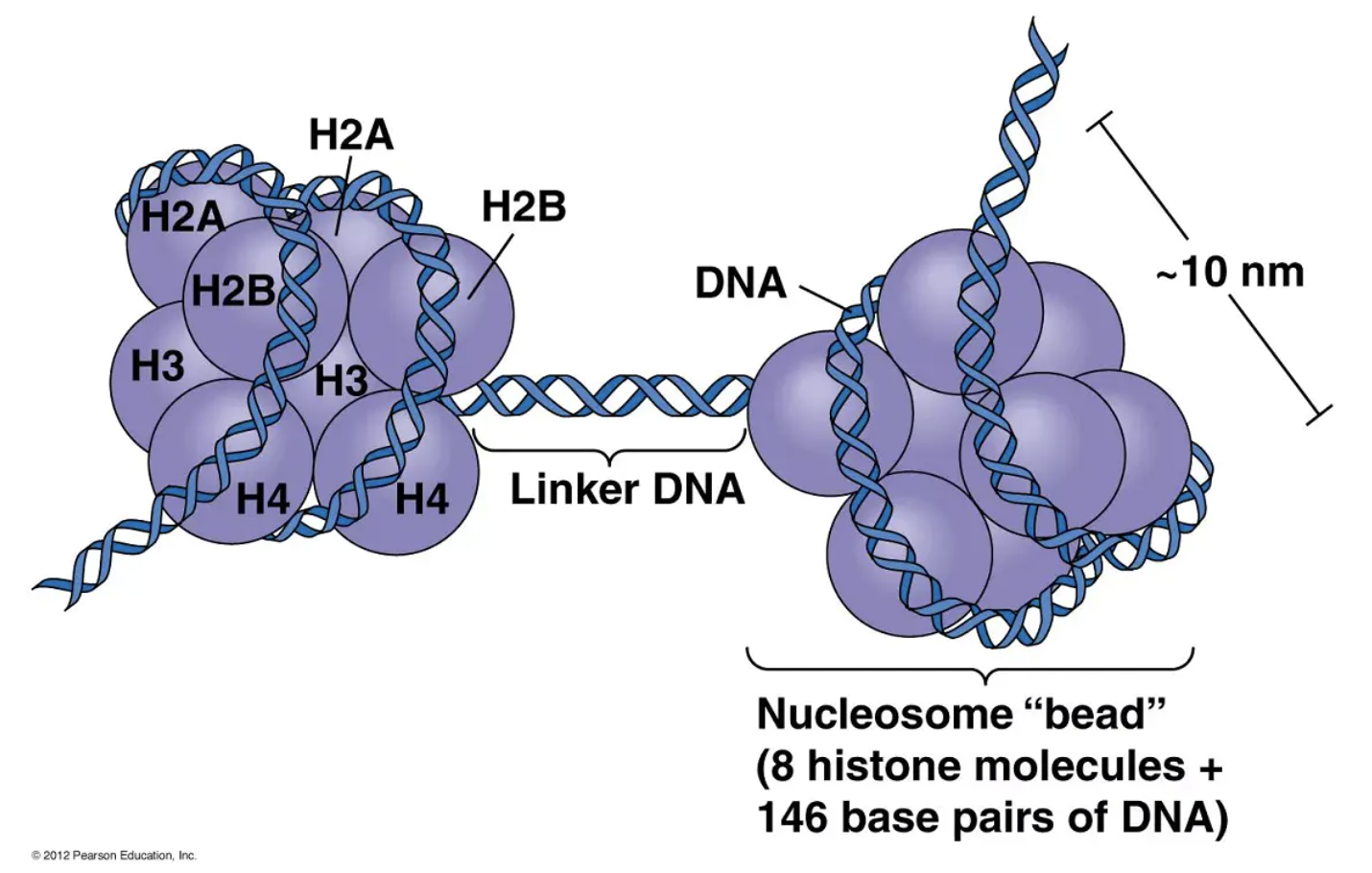
图6 核小体结构。(图片来源:https://www.jieandze1314.com/post/cnposts/191/)
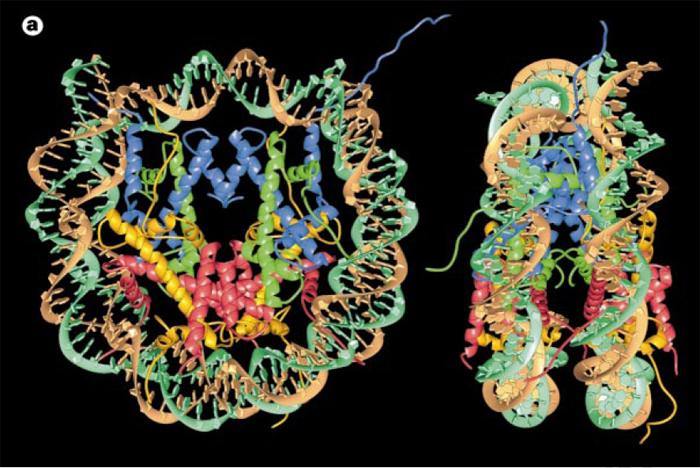
每个核小体的DNA数量是通过用一种切割DNA的酶(这种酶被称为DNA酶)来处理染色质来确定的。其中一种酶,微球菌核酸酶(MNase),具有重要的特性,即在切割包裹在八聚体周围的DNA之前,优先切割核小体之间的连接DNA。通过调节应用MNase后发生的切割量,有可能在每个连接DNA被裂解之前停止反应。此时,处理过的染色质将由单核小体、双核小体(通过连接物DNA连接)、三核小体等组成 (Hewish and Burgoyne, 1973)。如果MNase处理的染色质的DNA在凝胶上分离,就会出现许多条带,每个条带的长度是单核小体DNA的倍数(Noll, 1974)。对这一观察结果最简单的解释是,染色质具有基本的重复结构。晶体学家根据他们的数据构建的核小体模型如图7所示。DNA双螺旋的磷酸二酯骨架显示为棕色和绿松石色,而组蛋白显示为蓝色(H3)、绿色(H4)、黄色(H2A)和红色(H2B)。注意,只有真核生物(即具有细胞核和核膜的生物)才有核小体。原核生物,如细菌,则没有。
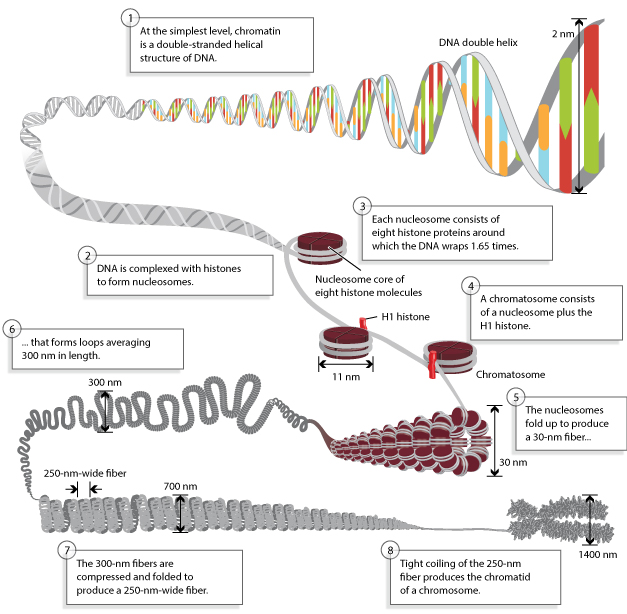
图7 染色体是由紧密缠绕在组蛋白上的DNA组成的(Annunziato A, 2008)。在组蛋白的帮助下,染色体DNA被包裹在微小的细胞核内。这些是带正电荷的蛋白质,它们强烈地粘附在带负电荷的DNA上,形成被称为核小体的复合物。每个核小体由8个组蛋白缠绕1.65圈的DNA组成。核小体折叠形成一个30nm的染色质纤维,形成平均300nm长的环。300nm的纤维被压缩和折叠,产生一个250nm宽的纤维,该纤维紧紧地盘绕在染色体的染色单体中。
图中的8个过程:
①在最简单的水平上,染色质是一个双链螺旋结构的DNA。
②DNA与组蛋白结合形成核小体。
③每个核小体由8个组蛋白组成,DNA围绕组蛋白包裹1.65圈。
④染色体由核小体加上H1组蛋白组成。
⑤核小体折叠形成30nm的染色质丝。
⑥30nm染色质丝平均每300nm盘曲成环。
⑦300nm的纤维被压缩和折叠,产生一个250nm宽的纤维。
⑧250nm纤维的紧密结合产生了一个染色体的染色单体。
Asensi-Fabado M A, Amtmann A, Perrella G. Plant responses to abiotic stress: the chromatin context of transcriptional regulation[J]. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms, 2017, 1860(1): 106-122.
Bradbury E M. KE Van Holde. Chromatin. Series in molecular biology. Springer‐Verlag, New York. 1988. 530 pp. $98.00[J]. 1989.
Chaitankar V, Karakülah G, Ratnapriya R, et al. Next generation sequencing technology and genomewide data analysis: Perspectives for retinal research[J]. Progress in retinal and eye research, 2016, 55: 1-31.
Chen X, Bhadauria V, Ma B. ChIP-seq: a powerful tool for studying protein–DNA interactions in plants[J]. Current Issues in Molecular Biology, 2018, 27(1): 171-180.
Choudhary C, Weinert B T, Nishida Y, et al. The growing landscape of lysine acetylation links metabolism and cell signalling[J]. Nature reviews Molecular cell biology, 2014, 15(8): 536-550.
Goldberg A D, Allis C D, Bernstein E. Epigenetics: a landscape takes shape[J]. Cell, 2007, 128(4): 635-638.
Hewish D R, Burgoyne L A. Chromatin sub-structure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease[J]. Biochemical and biophysical research communications, 1973, 52(2): 504-510.
Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression[J]. Nature Reviews Genetics, 2015, 16(2): 71-84.
Kim J M, Sasaki T, Ueda M, et al. Chromatin changes in response to drought, salinity, heat, and cold stresses in plants[J]. Frontiers in plant science, 2015, 6: 114.
Kinnaird A, Zhao S, Wellen K E, et al. Metabolic control of epigenetics in cancer[J]. Nature Reviews Cancer, 2016, 16(11): 694-707.
Kouzarides T. Chromatin modifications and their function[J]. Cell, 2007, 128(4): 693-705.
Luger K, Mäder A W, Richmond R K, et al. Crystal structure of the nucleosome core particle at 2.8 Å resolution[J]. Nature, 1997, 389(6648): 251-260.
Luo C, Lam E. Quantitatively profiling genome-wide patterns of histone modifications in Arabidopsis thaliana using ChIP-seq[M]//Plant Epigenetics and Epigenomics. Humana Press, Totowa, NJ, 2014: 177-193.
Luo M, Cheng K, Xu Y, et al. Plant responses to abiotic stress regulated by histone deacetylases[J]. Frontiers in plant science, 2017, 8: 2147.
Olins A L, Olins D E. Spheroid chromatin units (ν bodies)[J]. Science, 1974, 183(4122): 330-332.
Olins D E, Olins A L. Chromatin history: our view from the bridge[J]. Nature reviews Molecular cell biology, 2003, 4(10): 809-814.
Solomon M J, Larsen P L, Varshavsky A. Mapping proteinDNA interactions in vivo with formaldehyde: Evidence that histone H4 is retained on a highly transcribed gene[J]. Cell, 1988, 53(6): 937-947.
Thomas J O, Kornberg R D. An octamer of histones in chromatin and free in solution[J]. Proceedings of the National Academy of Sciences, 1975, 72(7): 2626-2630.
Wolffe, A. P. Chromatin: Structure and Function, 3rd ed. (San Diego, Academic, 1999)
Woodcock C L F, Safer J P, Stanchfield J E. Structural repeating units in chromatin: I. Evidence for their general occurrence[J]. Experimental cell research, 1976, 97(1): 101-110.
Zhu J Y, Sun Y, Wang Z Y. Genome-wide identification of transcription factor-binding sites in plants using chromatin immunoprecipitation followed by microarray (ChIP-chip) or sequencing (ChIP-seq)[M]//Plant Signalling Networks. Humana Press, 2011: 173-188.
