微生物组对于植物的营养和健康有着重要的影响,而宿主植物通过特定基因表达、代谢物、形态特征等塑造了与其共生的微生物组。然而,这些互作关系往往异常复杂,如何抽丝剥茧、鉴定塑造植物-微生物关系的关键因素,是当前研究的巨大挑战之一。最近几年,一些先锋研究尝试运用多组学(Multiomics)的手段,解析植物-微生物的互作关系,鉴定调控微生物组的植物遗传因素。

近日,浙江大学土水资源与研究所马斌课题组与茶叶所徐平课题组合作发文,通过综合分析100个茶树品种基因组,以及茶树叶际、根系内生、根际微生物群落,在宿主-微生物共生组学(Holo-omics)的理论框架下,运用微生物组全基因组关联分析(microbiome GWAS)等技术手段,揭示了植物遗传因子驱动微生物组在茶树不同部位的装配差异机制。相关成果题为“Host genetic determinants drive compartment-specific assembly of tea plant microbiomes”在线发表在植物学领域著名期刊《Plant Biotechnology Journal 》(生物一区Top,IF=13.263)。
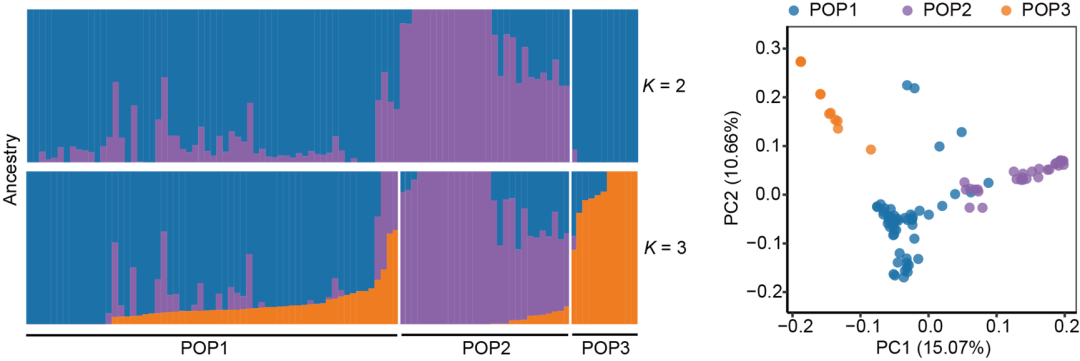
茶树是起源于我国的重要木本经济作物,有着悠久的栽培历史和广泛的分布区域。在长期的繁育中,茶树形成了丰富的种质资源,其多样化的形态和生理生化特征构成了不同风味茶饮的基础。研究人员首先对100个茶树品种的基因组进行了重测序,获得了~1600万个遗传位点的变异信息,并通过群体遗传分析厘清了品种间的亲缘关系。
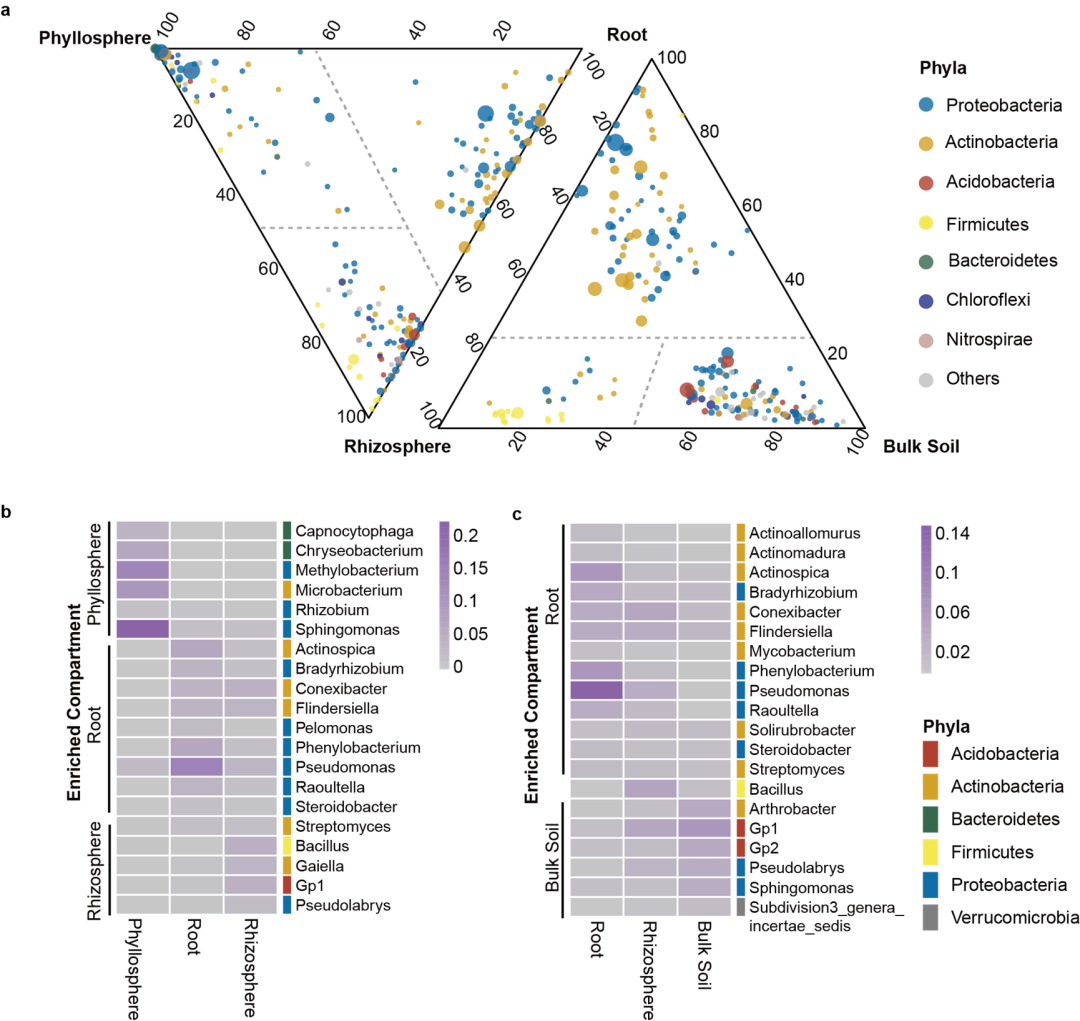
继而,研究者对这100个茶树品种的叶际、根内生、根际土壤、非根际土壤的1200个样品进行通过16S扩增子测序,分析了其细菌群落的组成和多样性,鉴定了不同植物区室微生物组的特异性装配。
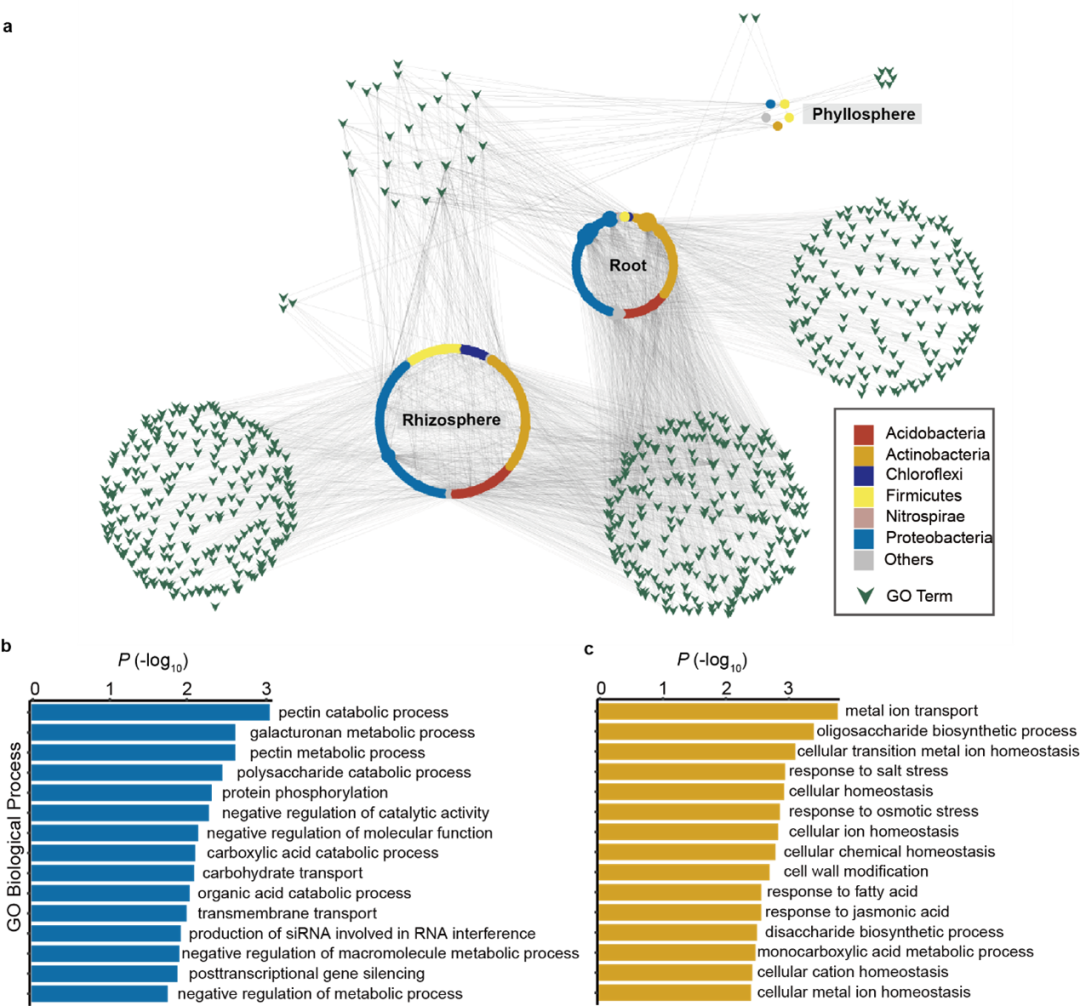
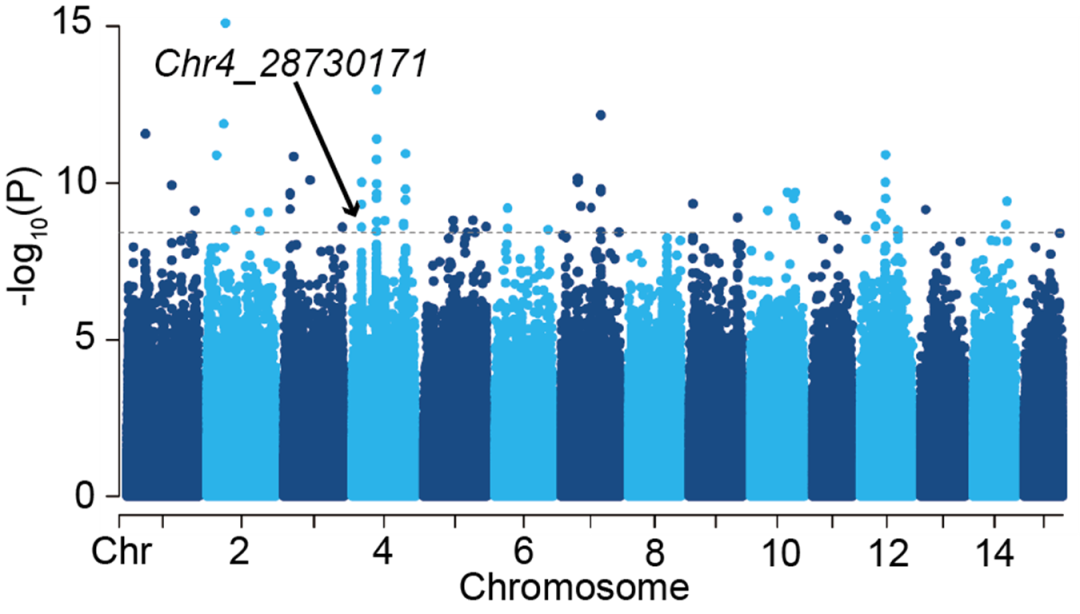
为了研究微生物群落组成与植物遗传变异的关系,研究者利用微生物组全基因组关联分析手段,将每一个微生物可操作分类单元(OTU)的相对丰度类比为植物的“性状”,与茶树遗传位点进行关联分析。在汇总了所有的关联结果后,研究者比较了与不同区室微生物组关联的遗传位点和候选基因。通过对这些基因的基因本体(GO)进行富集分析,发现与细胞壁、碳分解代谢相关的基因与根系内生微生物组的组成密切相关,而与离子、有机小分子代谢相关的基因在与根际微生物群落组成的关联中呈现富集趋势。这些结果表明,植物通过不同的代谢通路,影响不同区室微生物组的装配。研究中鉴定的候选基因,在调控茶树微生物组方面,具有一定的应用潜力。
进一步,研究者在群落水平上检验了微生物组β多样性与茶树遗传位点变异的关联。结果发现与细胞骨架有关的formin homology interacting protein 1基因,与叶际微生物群落的β多样性密切相关,表明其具有调控微生物组整体结构的潜力。
本研究通过跨学科合作,综合分析了宿主植物基因组和微生物组特征,揭示了茶树遗传多样性驱动微生物组在不同区室内的特异性组装机制,拓展了宿主-微生物共生组学的研究范畴,并为通过植物育种塑造与其共生的微生物组提供了理论基础。
浙江大学土水资源与研究所博士后谭向峰为论文第一作者,浙江大学马斌研究员、徐平副教授为论文通讯作者。浙江大学徐建明教授、王岳飞教授等参与指导该项研究。研究得到国家自然科学基金重大项目、青年基金以及浙江省自然科学基金的资助。